Pseudonitrosite FAQby RhodiumPseudonitrosite Synthesis, as Performed by Pugsley IntroductionThe hitherto pretty unknown pseudonitrosites are nitrogenous compounds which are formed by the action of N2O3 (an equimolar mixture of NO and NO2) upon etheral solutions of some unsaturated compounds, for example propenylbenzenes and styrenes. They were popular in the late 1800s and early 1900s, but has since then been forgotten(?). After around WWII, there is not a single mention of them in the literature, not even in such a reference work on the subject as Nitroalkenes - Conjugated Nitro Compounds [1]. The pseudonitrosites were discovered by Toennies around 1880, when he made the pseudonitrosite of anethol, but he failed to recognize the constitution of the compound, and mistakenly assigned it the structure of a vicinal ketoxime/nitrite. In the early 1890s, the Italian chemist Angelo Angeli experimented with different allyl- and propenylbenzenes, and again encountered the pseudonitrosites, but he couldn't either determine their exact structure, and with the methods used in analytical chemistry back then, this is understandable. In the early 1900s, the puzzle was solved by studies on styrene pseudonitrosite, and around this time Wallach and Wieland published reviews of pseudonitrosite chemistry [2-3]. Very little about pseudonitrosites can be found in Chemical Abstracts, so most pointers to earlier work must be retrieved from the references cited in those articles. In the 1930's, Victor Bruckner began to explore the possibility of using pseudonitrosites as an intermediate in the synthesis of substituted 1-phenyl-2-amino-propanols, and in his articles he also included improved syntheses of several pseudonitrosites from propenylbenzenes, as well as studies on the pseudonitrosites themselves. One of the most useful reactions he investigated was the basic hydrolysis of propenylbenzene pseudonitrosites, yielding the corresponding beta-nitro derivatives of the propenylbenzenes. This route offered several advantages over the known nitration of alkenes with tetranitromethane, which is expensive, toxic and explosive. In his papers on pseudonitrosites, Bruckner described several experiments with asarone [4], methylisoeugenol and isosafrole [5], isoeugenol [6] and anethole [7]. In the experimental part below, some freely translated parts of his work is included. Theory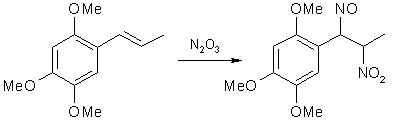
Figure 1: Formation of Asarone Pseudonitrosite Several methods for the synthesis of pseudonitrosites have been used, such as bubbling N2O3 through a solution of the propenylbenzenes in ether, slow addition of a NaNO2 solution to a solution of the propenylbenzene in glacial acetic acid (Toennies' method), as well as dripping a dilute solution of sulfuric acid into a two-phase solution of NaNO2 in water and a propenylbenzene in ether (Bruckner's method). 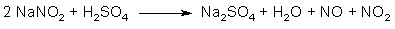
Figure 2: N2O3 from NaNO2 and H2SO4 The action of a strong acid (such as sulfuric acid) on sodium nitrite gives the sodium salt of the strong acid, as well as free nitrous acid (HNO2), which in turn breaks down to an equimolar mixture of NO and NO2 gas (see Fig. 2). This gas mixture is in equilibrium with dinitrogen trioxide: N2O3 <=> NO + NO2 At 25°C and normal pressure, only about 10% of the gas is in the form of N2O3, and the equilibrium mixture behaves just like as if it was a mixture of NO and NO2. But, for the sake of simplicity, the equilibrium mixture is commonly called N2O3, or dinitrogen trioxide. 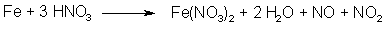
Figure 3: N2O3 from HNO3 and iron N2O3 can also be formed by the action of nitric acid (which in turn can be made by dissolving NaNO3 or KNO3 in conc H2SO4) on certain metals, for example iron (see Fig. 3). The more finely divided the metal, the faster generation of gas. But there is a downside to this method. In the beginning the nitric acid is concentrated, but as N2O3 is evolved, the solution becomes more and more dilute. There is a problem associated with this, the ratio of NO to NO2 isn't constant if the concentration of the acid changes. Concentrated acid gives excess NO2, and a dilute acid solution gives excess NO. To get maximum yields out of the treated propenylbenzene, the proportions between NO and NO2 should always be as close to 1:1 as possible, and only the sodium nitrite method will constantly produce a gas mixture with just those proportions. Producing dinitrogen trioxide with the nitrate method is therefore discouraged. This method does not work at all with asarone (only with other propenylbenzenes, such as isosafrole or anethole) according to Bruckner[4]. 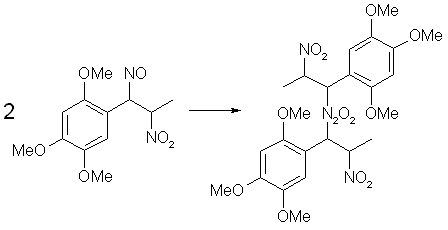
Figure 4: Dimerization of Asarone Pseudonitrosite The pseudonitrosites always dimerizes to the bis-pseudonitrosites, all of which are practically insoluble in water, alcohols and most common organic solvents, with the exception of warm chloroform or ethyl acetate, in which a blue-green solution is produced, consisting of the dissociated pseudonitrosite monomer. During the treatment of a propenylbenzene with N2O3, the intensely colored free monomer can be observed for a while before it dimerizes to its crystalline form. The colors of the crystalline pseudonitrosites varies, from the snow-white isosafrole or anethole derivatives to the canary-yellow one obtained from asarone. Upon storage, pseudonitrosites soon decompose with discoloration, releasing nitrogen oxides and hydrogen cyanide. At around 40°C decomposition takes place in less than one day. 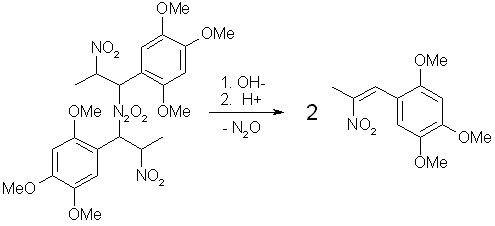
Figure 5: Hydrolysis of Asarone Pseudonitrosite Dimer Treatment of the pseudonitrosite dimer with four moles of an inorganic base (such as KOH in alcohol), produces two moles of potassium hyponitrite (K2N2O2) and two moles of the water-soluble potassium salt of the corresponding nitroalkene. Acidification of the potassium salt solution with dilute acid precipitates the water-insoluble phenyl-2-nitropropene as fine crystals, and the liberated hyponitrous acid breaks down into water and nitrous oxide (N2O), which can be observed as fine bubbles that is being evolved during this step. The reaction of a pseudonitrosite with acetic anhydride produces the alpha-acetoxy derivative with release of N2O, and after reduction of the nitro group this could be hydrolyzed to ring substituted phenylpropanolamine derivatives. Basic hydrolysis of the alpha-acetoxy derivative also produces the phenyl-2-nitropropene. Reaction of pseudonitrosites with primary amines yields secondary alpha-amino derivatives. Experimental ProceduresAll these procedures are presented for informational purposes only. These procedures should not be carried out without adequate precautions taken. In most of the procedures highly toxic fumes of nitrogen oxides can evolve, which are irritating on mucous membranes and can destroy lung tissue. Use only with good ventilation. The author assumes no responsibility for any damage or legal consequences resulting from misuse of this information. Pseudonitrosites [4,5,7]A solution of 1 mole (69 grams) of sodium nitrite in 100 ml water was prepared in a 500 ml flask, and a solution of 0.1 moles of a freshly distilled propenylbenzene (20.8g asarone, 16.2g isosafrole or 14.8g anethole) in 150 ml of diethyl ether was added. During a period of 3-4 hours, a 20% solution of H2SO4 (prepared by cautiously adding 0.5 moles (49 grams) of conc. sulfuric acid to 200 ml of H2O) was added dropwise with magnetic stirring, preferably through a pressure- equalized addition funnel. Watch the addition rate so that the temperature of the two-phase solution doesn't rise over room temp, and this solution can preferably be cooled during the addition In the beginning of the addition of acid, monomeric pseudonitrosite can be seen in the etheral layer. The monomer derived from asarone is greenish, anethol blueish and isosafrole yellow. If oxygen gets into the system during the addition, formed NO oxidizes to brown NO2 gas, which can lower the yield of pseudonitrosite. After the addition, the solution was allowed to stir for an hour or two, whereafter the solution was filtered with suction, and the precipitate washed with 50ml each of water, denatured alcohol and finally ether. After sucked as dry as possible, the pseudonitrosite crystals was allowed to air dry on a filter paper.
Pseudonitrosites cannot be stored, as they begin to decompose within hours at room temperature with the evolution of nitrogen oxides, and should be further processed to for example the very useful phenyl-2-nitropropenes, which upon reduction can yield phenylacetones and amphetamine derivatives. Phenyl-2-nitropropenes [4,5,7]0.1 mole of the corresponding propenylbenzene pseudonitrosites (28.5g asarone, 23.8g isosafrole or 22.4g anethole) was dissolved in 150ml 8% alcoholic KOH with shaking, and possibly light heating (not over 30°C, especially not in the case of asarone pseudonitrosite, or there is a risk of decomposition (to the aldehyde, amongst other things). Caution: Foaming with evolution of N2O will occur. The cloudy solution was suction filtered, and the filtrate was poured onto 100 grams of crushed ice. The solution was acidified with dilute HCl and was stirred occasionally until the ice had melted. The precipitate of nitropropene was filtered off, washed with a little cold water and air dried.
[*]2-Nitroasarone exists in two modifications, yellow or red prisms, and depending on concentration and precipitation speed, one often gets a mixture of both species. They both melt at 101°C, the red form transforming itself to the yellow form at 90°C. References[1] V. V. Perekalin, E. S. Lipina, V. M. Berestovitskaya, D. A. Erefmov, Nitroalkenes (Wiley, 1994)[2] H. Wieland, Zur Kenntniss der Pseudonitrosite, Ann. 329, 225-268 (1903) [3] O. Wallach, Ueber die Additionsproducte von N2O3 und von NOCl, Ann. 332, 305-336 (1904) [4] V. Bruckner, Ueber das Pseudonitrosit des Asarons, J. Prakt. Chem., 138, 268-274 (1933) [5] V. Bruckner, Ueber die Verwendung der Pseudo-nitrosite , Ann. 518, 226-244 (1935) [6] V. Bruckner, Ueber die Verwendung der Pseudo-nitrosite II, J. Prakt. Chem. 143, 287-297 (1935) [7] V. Bruckner, Ueber die Verwendung der Pseudo-nitrosite III, J. Prakt. Chem. 148, 117-125 (1937) |