Synthesis of Propenylbenzenes from Propiophenonesby PsychokittyAnyway, I may have some good news for you guys. A friend of mine recently sent me a detailed lab experiment that was once a part of the Organic Chemistry program at some college in the US. In it is the synthesis of 4-methoxypropenyl-benzene from 4-methoxypropiophenone. My friend had apparantly e-mailed the chemistry teacher from that college who wrote up the experiment and he gave him a little more information than what was available in lab note book. Here is that information: Preparation of the Propiophenone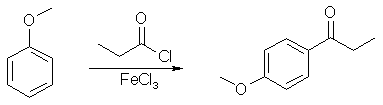
In a small Erlenmeyer flask, weigh out 2.16 g. of anisole and dissolve it in 5 ml of methylene chloride. Cover and set aside. Quickly weigh out and transfer 3.3 g of FeCl3 into a 50 mL round bottom flask with a magnetic stirrer and cover. With stirring, first add 25 mL of methylene chloride, then slowly add 2.0 mL of propionyl chloride (careful, smelly and toxic. Use the glass pipette in the hood). Put a Claisen head adapter on the round bottom flask, then place a separatory flask and a bent vacuum adapter on the Claisen head (see the drawing below). Transfer the anisole solution to a separatory funnel (the small one), which will serve as an addition funnel. Put a stopper in the bent vacuum adapter, run a hose to an inverted liquid funnel which is just above a beaker full of water (this will scub the HCl vapors). With stirring, add the anisole solution over a 2-4 min period (about 1 drop per 3-5 sec). The reaction is exothermic and evolves HCl, so you must be very careful not to add the solution too fast. After the addition, let it stir for another 20 min. Add 5 mL of water dropwise (use the sep funnel again) to quench the reaction and stir for 5 min. Transfer the contents of the flask with water (50 mL) and methylene chloride (10 mL). Shake vigorously, then separate the organic layer from the aqueous layer. Wash the aqueous layer with another 10 mL of methylene chloride, combine the organic layers, and wash the organic layer with 50 mL of 0.25 M NaOH. Dry the organic layer with Na2SO4, add 0.25 g of activated charcoal (add more if it does not seem sufficient for the removal of most of the colored impurities), and swirl for several minutes. Filter the mixture through a bed of Celite (vacuum filtration). Use a little methylene chloride to wash the filter cake. Put the organic solution in a tarred beaker in the back of the hood until next week. Obtain a weight, IR and NMR. Reduction of the Ketone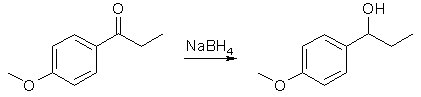
Dissolve the ketone (2 g) in ethanol (10 mL) in a 50 mL round bottom flask with a magnetic stirrer. Add NaBH4 (0.4 g) over a period of 15 min. The reaction may be mildly exothermic, so do not add the hydride reagent too fast. After stirring for 15 min, add 10 mL of water, heat to reflux for 5 min, and cool to room temperature (this destroys most of the excess sodium borohydride). Wash the reaction mixture into a separatory funnel with additional water (25 mL) and diethyl ether (50 mL). Shake, separate the organic layer and wash the aqueous layer again with ether (25 mL). Combine the organic layers, wash with water (25 mL), separate, and dry the organic layer over Na2SO4. Allow the organic solution to evaporate in the back of the hood in a tared flask until next week. After weighing your product, obtain an IR and NMR. Dehydration of the Alcohol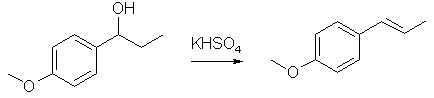
In a 50 mL round bottom flask, add 0.5 g of alcohol, a magnetic stirrer, and 25 mL of a 1.5 M solution of KHSO4. Set up a distillation apparatus on top of your round bottom flask (your lab notebook has a schematic for distillation set-ups) and distill off approximately 20 mL of liquid. This procedure not only drives the dehydration reaction to completion but it also purifies your product by steam distillation. Transfer the distillate to a separatory funnel, extract the aqueous phase twice with ether (2 tms 15 mL), dry the ether solution over Na2SO4, and evaporate the ether solution in a tared beaker in the hood in a warm sand bath (if you can't deal with the sample this week, make sure you cover up the flask with aluminum foil so that the anethole does not evaporate; it is fairly volatile). As before, get a weight, IR and NMR. So what do you think? My guess is that anisole could be replaced easily with benzodioxole or even benzene. Of course, if you already have propiophenone, you could jump directly to the second step. As for the very last part of the reaction, I would substitute heating the P-1-Pol with dry KHSO4 rather than use the aqueous KHSO4 solution like the proceedure describes. Also, if one cared to form the benzoyl acetate first from the propiophenone and then reduce to the diol, I suppose step two could be used to effect this reaction as well -- in lieu of the traditional and expensive catalytic reduction using Pt/C or whatever. For more information on this specific reaction, refere to the following citation: "Iron (III) Chloride as a Lewis Acid in the Friedel-Crafts Acylation Reaction" taken from Journal of Chemical Education -- Ahhh, SHIT! The article is right in front of me, but it doesn't indicate which volume it comes from. Oh, well. I know where to find the reference again. I'll get it to you guys soon. BTW, according to my friend, the professor indicated that only the last part tended to produce low yields (by student, 30-50%; by the professor, 50-70%) and that the first and second steps were high-yielding. If anyone wants another high yielding equally simple alternative to the first step in the above reaction, refer to "2,5-dimethoxy-n-propyl- either phenethylamine or amphetamine" in PIHKAL. There he uses 1,4-dimethoxybenzene in Friedel-Crafts fashion using AlCl3 and propionyl chloride in methylene chloride to form in about an hour the desired propiophenone. This method is probably easily adapted to using both benzodioxole and benzene itself. |