2 PdCl2 + 8 PPh3 + 5 NH2NH2·H2O → 2 Pd(PPh3)4 + 4 NH2NH2·HCl + N2 + 5 H2O
Preparations of phosphine and phosphite complexes of palladium(0) have been reported to result from reduction of palladium(II) complexes in the presence of the desired ligand1-5. The products are generally formulated as PdL4-n (where n = 0, 1), depending on the nature and amount of the ligand used. A related complex, [Pd(PPh3)2]n has also been reported6.
Although this preparation is similar in concept to these previous ones, advantage is gained in being able to obtain a high yield of Pd(PPh3)4 in one step from palladium dichloride.
Procedure
A mixture of palladium dichloride (17.72 g, 0.10 mol), triphenylphosphine (131 g, 0.50 mol), and 1200 mL of dimethyl sulfoxide is placed in a single-necked, 2-L, round-bottomed flask equipped with a magnetic stirring bar and a dual-outlet adapter. A rubber septum and a vacuum nitrogen system are connected to the outlets. The system is then placed under nitrogen with provision made for pressure relief through a mercury bubbler. The yellow mixture is heated by means of an oil bath with stirring until complete solution occurs (~140°C). The bath is then taken away, and the solution is rapidly stirred for approximately 15 min. Hydrazine hydrate (20 g, 0.40 mol) is then rapidly added over approximately I min from a hypodermic syringe. A vigorous reaction takes place with evolution of nitrogen. The dark solution is then immediately cooled with a water bath; crystallization begins to occur at ~125°C. At this point the mixture is allowed to cool without external cooling. After the mixture has reached room temperature it is filtered under nitrogen on a coarse, sintered-glass funnel. The solid is washed successively with two 50-mL portions of ethanol and two 50-mL portions of diethyl ether. The product is dried by passing a slow stream of nitrogen through the funnel overnight. The resulting yellow crystalline product weighs 103.5-108.5 g (90-94% yield)Note 1.
A melting point determinationNote 2 on a sample in a sealed capillary tube under nitrogen gave a decomposition point of 116°C (uncorrected). This compares with a similar determination (115°C) performed on the product prepared by the method of Malatesta and Angoletta1.
Properties
The Pd(PPh3)4 complex obtained by this procedure is a yellow, crystalline material possessing moderate solubilities in benzene (50 g L-1), dichloromethane, and chloroform. The compound is less soluble in acetone, tetrahydrofuran and acetonitrile. Saturated hydrocarbon solvents give no evidence of solution. Although the complex may be handled in air, it is best stored under nitrogen to ensure its purity.
In benzene, molecular-weight measurements suggest substantial dissociation1,4:
Pd(PPh3)4 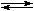 Pd(PPh3)4-n + n PPh3
Pd(PPh3)4-n + n PPh3
Solutions of the complex in benzene rapidly absorb molecular oxygen giving an insoluble, green oxygen complex, Pd(PPh3)2O27. This oxygen complex has been implicated as an intermediate in a catalytic oxidation of phosphines2,8.
Related displacements with acetylenes9 and electrophilic olefins6 have been reported to give complexes formulated as [Pd(PPh3)2 (olefin or acetylene)]. Also, oxidative additions of alkyl and aryl halides have been shown to occur giving palladium(II) complexes, Pd(PPh3)2(R)Cl10. As a catalyst, the complex has been shown capable of dimerizing butadiene to give 1,3,7-octatriene11.
Notes
- The checkers worked on one-third of the stated scale, obtaining a yield of 37.4 g (97%).
- The checkers report that decomposition temperature does not appear to be a good criterion of identity or purity since it is not very reproducible.