Abstract
The synthesis of the title compound is intended to proceed from the precursor para-flurobenzaldehyde. Using the following method, the nitroalcohol is to be formed. A 1:1,2:1 molar ratio of benzaldehyde : nitroethane : triethylamine will be used in a proper solvent. The product is to be reduced with Zn/formic acid to the aminoalcohol, which is to be reacted with sodiumcyanate, to make the carbamyl-intermediate. This may or may not be isolated and then further reacted with hydrochlorid acid, to form the oxazoline ring. The product is supposed to be non-neurotoxic.
Experimental
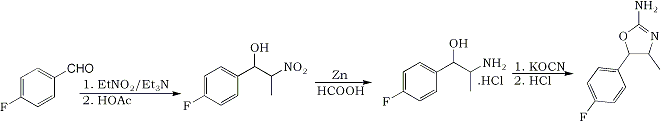
Condensation:
10 g (80.6 mmol) of para-fluorobenzaldehyde (99%+ purity, 124.11 g/mole) was mixed with 15 mL’s of methanol in a 100 mL Erlenmeyer flask, and put in the fridge and refrigerated till the temperature was -10°C. Fortunately the benzaldehyde was soluble at these low temperatures, which means that the reaction can be conducted as originally intended. In a separate beaker, 7.3 g (96.7 mmol) of nitroethane (75 g/mole) was mixed with 8.14 g (80.6 mmol) of triethylamine (99%+ purity, 101 g/mol). This was also cooled to –10°C. Once the mixture was cooled satisfyingly, the contents from the beaker with nitroethane/triethylamine was poured into the Erlenmeyer flask with the fluoro-benzaldehyde. This was swirled a few times, and put back in the freezer. Once every 30 min’s it was taken out and swirled. The color of the mixture went from totally clear to yellowish over the course of the 2½ hours, which the reaction was allowed to run. Once the reaction was complete (2½ hours), the amine was quenched with an 1.1 equimolar amount of acetic acid, while the reaction mixture was still cool. It is very important to quench the reaction while it is still very cold, as the isomers will reach an equilibrium once the temperature rises, if there still is active catalyst present. Most of the solvent was stripped under vacuum, and the remains where dissolved in DCM and washed two times with water and once with brine. The DCM was stripped, leaving behind about 15 g of the crude nitroalcohol.
Zn/Formic acid reduction:
The nitroalcohol was reduced in the usual manner, by Zn/formic acid. This method was used, with the following amounts:
- 15 g crude nitroalcohol
- 100 mL methanol
- 18.6 g Zinc Powder
- 70 mL of formic acid
After filtering the reaction mixture, a slightly reddish liquid remained. Most of the solvent was stripped, and the remains where dissolved in 100 mL water and acidified with hydrochloric acid to pH 2 (for some reason it was almost neutral, even though a huge excess of formic acid was used; maybe the zinc ate most of it?). The mixture was nearly pink at this point. This was washed twice with DCM to give a very pale red-tinted solution. Upon basification of this mixture, a lot of inorganic salts precipitated. This has probably been some Zinc salts of some sort. The whole lot was extracted once with chloroform and twice with DCM. After the first chloroform extraction and addition of DCM, the whole lot was filtered to remove the inorganic salts. The combined organic phases where stripped of solvents and left 7.6 g of para-fluoro-norephedrine (42.5 mmol) freebase as a pale yellow liquid.
Reaction with cyanate:
The para-fluoro-norephedrine was poured into 70 mL of water and titrated with hydrochloric acid, until weakly acidic, where after the while lot dissolved. 3.5 g (44 mmol) of KOCN was added to this in one portion. The mixtures was refluxed in an oil bath for 2½ hour. After this time a clear oil was present at the top of the refluxing water. After completion of the reaction, the mixture was put in the freezer in order to isolate the carbamoyl intermediate. A slightly reddish oil precipitated on the bottom of the flask, but no crystals formed. It was decided, simply to do the reaction as a one pot synth.
Formation of the oxazoline ring:
The reaction mixture was mixed with a 3 times molar excess of hydrochloric acid and refluxed for 2½ hour. After this time, the flask was removed from the oil-bath and cooled. No oil was present anymore, so a reaction has definately occurred. At 40°C, sodium carbonate as a 20% solution was added to part of the solution. The mixture turned opaque and white upon basification, but no real formation of crystals could be noted right away.
Post reaction workup:
The post reaction mixture was washed twice with 30 mL of DCM to remove any amide present. After discarding the DCM the water phase was basified with sodium carbonate and a little sodium hydroxide. The solution turned nice and cloudy . This was extracted with 3x30 mL DCM. The DCM was evaporated under reduced pressure and 2.6 g of slighly yellow oil remained which can be converted to preferred salt or can be used as freebase. The oil was negative on marquis test.
The overall yield from the fluorobenzaldehyde was 19%, the yield of the first step was 53% and the last was 35%. It's somewhat on the low side, but if this compound turns out to be very potent it's not too bad. The synthesis is very easy and it's obtainable chemicals that are used.
4'-fluoro-(4-methylaminorex) has greater affinity for 5-HT uptake than 4-methylaminorex, which indicates it's weaker stimulant properties. Dosage range is around 20-40mg.