Abstract
Stereospecific synthesis of the racemic threo isomers of 2-nitro-1-phenylpropanols by reacting a benzaldehyde derivative with nitroalkane in the presence of a tertiary amine and reducing 2-nitro-1-phenylpropanols with, for example, lithium aluminum hydride to 2-amino-1-phenylpropanols is described. Also described are phase transfer resolution of racemic mixtures of 2-amino-1-phenylpropanol and its derivatives into their optically pure isomers by reacting a racemic mixture with the mono alkali metal salt of a tartaric acid ester in a two phase system of a hydrocarbon and water. The specification further describes therapeutically useful optically pure isomers of threo-2-amino-1-(dialkoxy or alkoxy) phenylpropanols and acid addition salts thereof.
Prior Art
Mixed stereoisomers of 2-amino-1-phenylpropanol can be readily prepared by reacting benzaldehyde with nitroethane in the presence of an alkaline catalyst to produce 2- nitro-1-phenylpropanol which is then reduced to the amine. E.g., Hoover and Hass, Journal of Organic Chemistry, 12, 506, (1947). This reaction gives excellent yields at low cost. However, separating the stereoisomers produced by this reaction has not been satisfactory. As a result, heretofore, the only practical stereospecific synthesis of 2-amino-1-phenylpropanols involves the reduction of propiophenone derivatives to the racemic erythro diastereoisomers and fractional crystallization of the amine salt of an optically active acid.
The DL-erythro forms can be readily obtained from the appropriate propiophenone derivatives or by inversion of DL-threo derivatives. Since the pharmacological properties of the D and L isomers differ, it is desirable to separate the two, resulting in maximum therapeutic utility. While such potentially useful products can also be prepared, the difficulty of separating, i.e. "resolution", the isomers has prevented their development.
Specifically, the base is converted to a salt of an optically active acid. For example, the DL-base is reacted with a D-acid. This results in the formation of a mixture of D-base-D-acid and L-base-D-acid. These two salts differ in solubility. When the mixture is cooled and allowed to stand, the D-base-D-acid, typically being the less soluble, precipitates out of the solution first. By removing the precipitate at the appropriate time, the collected precipitate is largely the D-base-D-acid, while the L- base-D-acid remains in solution.
While this results in purification of the optical isomers to some extent, substantial impurities often remain. These impurities are removed to some degree by fractional crystallization. The "pure" D and L bases are then liberated by adding a sufficient amount of an alkali to a solution of the "pure" salt to produce a pH above 7.
The above process is very tedious, time consuming, and inefficient. In addition, a base with the same configuration as the resolving acid is more easily separated and purified than the opposite enantiomer. Consequently, in practice, the desired isomeric base must be matched with an optically active acid of the same configuration to achieve maximum yields.
Description Of Invention
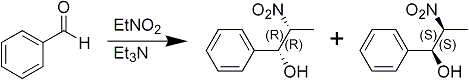
It has been discovered that if a benzaldehyde derivative is reacted with a nitroalkane in the presence of a tertiary amine, the nitroalcohol formed is of a threo configuration rather than a mixture of threo and erythro isomers which results when sodium hydroxide is used as the catalyst. The reaction is best conducted in an aqueous aliphatic alcohol.
The tertiary amine employed must be free of primary and secondary amines which interfere with the reaction. These can be removed from the commercial products by refluxing with acetic or phthalic anhydride followed by distillation.
In a preferred embodiment of the synthetic method of the present invention, a benzaldehyde derivative is reacted with nitroethane in the presence of triethylamine in aqueous ethanol. The mixture is allowed to react at room temperature for twenty-four hours. The mixture is then acidified with an organic acid since mineral acids tend to promote decomposition, as does heat. Excess solvents and reactants are evaporated and the nitroalcohol extracted.
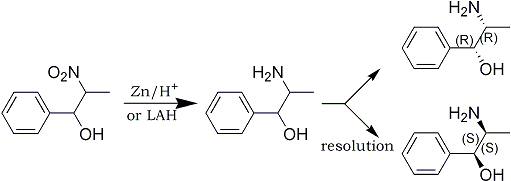
The nitroalcohol can easily be reduced by catalytic hydrogenation or conventional reducing agents such as zinc and acid. Lithium aluminum hydride has been found to work exceptionally well. The racemic threo isomers can readily be inverted to the racemic erythro isomers by reaction with acetic anhydride and thionyl chloride and hydrolysis. N-methyl derivatives can be prepared by adding an equimolecular amount of aqueous formaldehyde to the primary amine and reducing the Schiff base The use of a primary aliphatic alkylamine results in the formation of the dehydration product of the nitroalcohol - the beta-nitrostyrene. By employing a secondary or tertiary amine in the presence of water, nitrostyrene formation is completely eliminated. The use of a secondary or tertiary amine prevents the formation of a Schiff base amine, the necessary intermediary for nitrostyrene formation.
Instead of the usual fractional crystallization of the amine-acid salt, the method of resolving stereo isomers of the present invention uses a novel phase transfer resolution. It has been discovered that if the amine base is rapidly stirred with a mono alkali metal salt of a tartaric acid ester in a two-phase system of a hydrocarbon and water, a rapid and efficient resolution can be achieved.
In a practical embodiment of the resolution method of the present invention, a DL-2-amino-1-phenylpropanol derivative in dichloromethane is combined and stirred with dibenzoyltartaric acid in water, and aqueous sodium hydroxide for from about one to three hours. The reaction mixture is then allowed to stand for the about same length of time. The dichloromethane phase is separated and dried over anhydrous magnesium sulfate. Evaporation gives the L-threo isomer in nearly quantitative yield. The aqueous phase is made alkaline with ammonia and extracted with dichloromethane. The dichloromethane extract is dried over anhydrous magnesium sulfate and evaporated to give the D-threo isomer in nearly quantitative yield.
Experimental
Example 1
A racemic mixture of threo nitroalcohols was prepared by combining freshly distilled benzaldehyde (1 mole), nitroethane (2.5 moles), and triethylamine (0.05 mole) in ethanol (150 ml.) with water (75 ml). This mixture was allowed to stand at room temperature in the dark for twenty-four hours. The mixture was then ice-cooled and acetic acid (0.05 mole) was added to the reaction mixture. Alcohol and excess nitroethane were evaporated (vacuum). Water (75 ml) was added and the nitro alcohol extracted with ethyl acetate, dried over anhydrous sodium sulfate and the solvent evaporated (vacuum) to give the product, a viscous oil (yield 70-80% based on the benzaldehyde).
Example 2
The nitro alcohols were reduced by two methods, a zinc and acid method and a lithium aluminum hydride method as described below:
(A) Zinc and Acid
Hydrochloric acid (4 moles) is added (with stirring) to a mixture of nitroalcohol (1 mole), zinc dust (4 moles), and 400 ml. of 95% ethanol. The acid is added at such a rate that the temperature remains at 45 degrees or below (several hours are usually required). Stirring is continued for 1-2 hours after completing the addition. The acid solution is extracted with ether to remove non-basic materials. Excess NaOH solution is then added and the free base extracted with ether. The ether solution is dried (MgSO4) evaporated, and the product distilled or crystallized in the usual manner (70-80% yield).
(B) Lithium Aluminum Hydride ("LAH")
A solution of the nitroalcohol (1 mole) in tetrahydrofuran (400 ml) is added to a solution of lithium aluminum hydride (4 moles) in tetrahydrofuran (500 ml) with rapid stirring and cooling (as necessary) to maintain gentle reflux. After the addition is complete, the mixture is refluxed for an additional 2 hours.
Water is added to neutralize excess LAH (1 liter) and the product extracted with benzene. The benzene extract is dried (MgSO4) and the benzene evaporated. The resulting product is purified by either distillation or crystallization (80-90% yield).
Example 3
The reaction mixture of reduced nitro alcohols was resolved into optically pure isomers by the following process.
A mixture of a DL-threo-2-amino-1-phenylpropanol (1 mole) in dichloromethane (600 ml), dibenzoyltartaric acid (0.5 mole) in distilled water (30 ml), and sodium hydroxide (0.5 mole) in distilled water (50 ml) is stirred rapidly for two hours and allowed to stand for two hours. The dichloromethane phase is separated using a separating funnel over anhydrous magnesium sulfate. Rotary evaporation of the dichloromethane phase gives the L-threo isomer in nearly quantitative yield.
The aqueous phase is made alkaline with ammonia to pH 13 and extracted with
dichloromethane. The dichloromethane extract is dried over anhydrous magnesium
sulfate and evaporated to give the D-threo isomer in nearly quantitative
yield. The enantiomeric purity of the products is 96-99% based on GLC analysis
of the D or L-  -methoxy--
trifluromethylphenylacetamide (MTPA) derivatives.
-methoxy--
trifluromethylphenylacetamide (MTPA) derivatives.