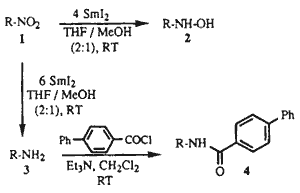
The reaction of SmI2 with nitroarenes gives the corresponding anilines1. However, to our knowledge there are no literature precedents for the reduction of alkyl nitro compounds to either alkyl hydroxylamines or alkyl amines by SmI2. During the course of our studies using SmI2 as a reducing agent2, we found that in the presence of MeOH as a proton source, primary, secondary, and tertiary alkyl nitro compounds were reduced by SmI2 to either hydroxylamines or amines depending upon reaction conditions.
Table 1.
Reduction of Nitroalkanes
With 4 Equivalents SmI2
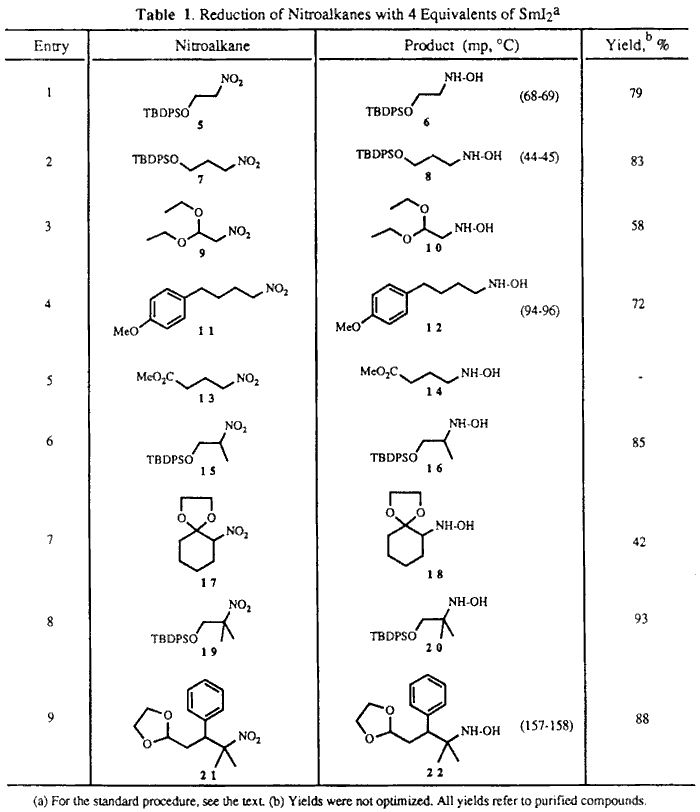
Table I shows that reductions to the hydroxylamines proceeded
well with either primary, secondary, or tertiary nitroalkanes. In general,
reduction of the nitro group with 4 molar equivalents of SmI2 in
THF/MeOH (2:1) proceeded smoothly in less than five minutes and simple workup
of the reaction mixture alIowed isolation of the corresponding hydroxylamines
in good yields. The TBDPS group was well tolerated (entries 1,
2, 6, and 8). Yields appeared
Iower for substrates 93 and
174 containing an acetal or
a ketal functionality at the  -position
(entries 3 and 7). However, the alkyl nitro
compound 215 containing an
acetal at the
-position
(entries 3 and 7). However, the alkyl nitro
compound 215 containing an
acetal at the ![]() -position
gave the corresponding hydroxylamine in 88% yield (entry 9).
In the case of the nitro ester 13, the expected hydroxylamine
was not formed, and only a complex mixture was obtained (entry 5).
-position
gave the corresponding hydroxylamine in 88% yield (entry 9).
In the case of the nitro ester 13, the expected hydroxylamine
was not formed, and only a complex mixture was obtained (entry 5).
Experimental
A typical procedure for the SmI2 reductions of Table 1 follows. To a stirred solution of freshly prepared6 SmI2 (4.0 mmol) in THF (30 mL) was rapidly added a solution of the nitro compound 5 (1.0 mmol) in a 2:1 mixture of THF/MeOH (6 mL). The reaction mixture was stirred at RT for 3 minutes, poured into a 10% solution of Na2S2O3 (30 mL) and extracted with EtOAc several times. The residue was chromatographed over silica gel (EtOAc) to give 6 in 79% yield.7,8
When the reductions were performed with 6 molar equivalents of SmI2 at RT for several hours (see Table 2), the nitroalkanes were cleanly transformed into the corresponding primary amines, identified in this study as the corresponding 4-phenylbenzamides. Thus, to a stirred solution of SmI2 (4.2 mmol) in THF (30 mL) was added a solution of nitroalkane 15 (0.7 mmol) in a 2:1 mixture of THF/MeOH (6 mL). The reaction mixture was stirred for 8 h to give after workup as above the crude amine 29. This was dissolved in CH2Cl2 (15 mL) and treated with Et3N (0.5 mL), then with 4-phenylbenzoyl chloride (2.3 mmol), and stirred at RT overnight. The reaction mixture was diluted with EtOAc and H2O, the organic phase separated, and the aqueous phase extracted with EtOAc several times. The residue was chromatographed over silica gel (hexane/EtOAc, 85:15) to give 30 in 79% yield.9
Table 2.
Reduction of Nitroalkanes
With 6 Equivalents SmI2
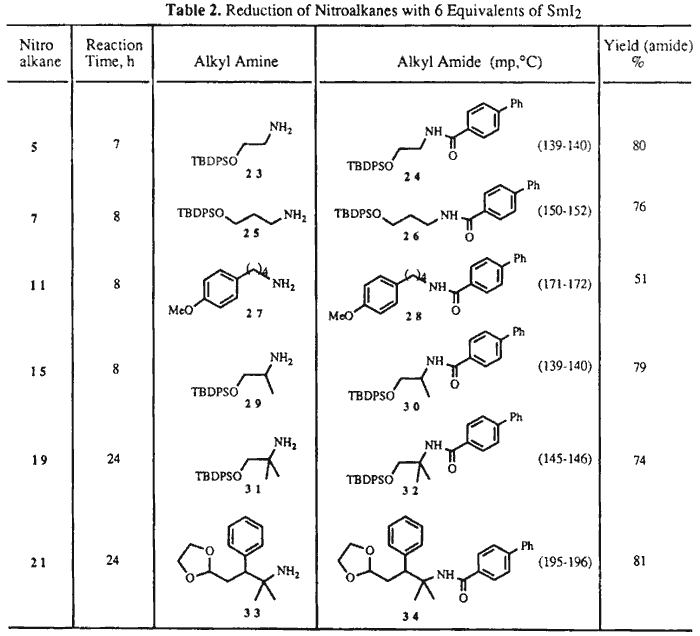
When 3-nitro acetal 9 or 3-nitro ketal 17 were reacted with 6 equivalents of SmI2 under the above reaction conditions, less than 20% of a complex mixture of unidentified products was isolated. Rapid decomposition of the primary reduction products could be observed under the reaction conditions.
Thus, we have demonstrated that in the presence of MeOH as a proton source, using 4 or 6 molar equivalents of SmI2 and by controlling the reaction time, a variety of nitroalkanes are cleanly reduced to either hydroxylamines or amines in moderate to good yields. This mild method offers exceptional simplicity and convenience in workup and compares very favorably to alternative procedures for laboratory syntheses of alkyl hydroxylamines or alkyl amines10.