Reductive Amination of MDP2P using Sodium Cyanoborohydride (NaBH3CN)by John Payne[ Back to the Chemistry Archive ] Sodium cyanoborohydride is most useful for the synthesis of MDMA and methamphetamine, less so for MDA. However, Uncle Fester says that sodium cyanoborohydride gives him poor yields and he dismisses its usefulness. Uncle Fester is wrong on this count and on other points. Sodium cyanoborohydride reductive amination of phenylacetone or 3,4MDP2P can give up to 98% yield in very large batches with no side reactions. Alexander Shulgin in PiHKAL discusses the use of sodium cyanoborohydride reductive amination but only for very small batches. In fairness to both authors it should be said that Fester is a good read but is often wrong, whereas Shulgin is a difficult read but is often deficient. Uncle Fester presents the use of activated aluminium for reductive amination and so does Shulgin. Aluminium may be the best way to go, but since I have not yet had the opportunity to try it, I am not in a position to comment. Let us start with theory. Why do reductive amination at all? Many poor thinkers advocate the bromination of safrole with hydrobromic acid followed by amination with methylamine. What could be simpler? What could be more stupid? Chemists have been synthesising various amphetamines for nearly one hundred years now using various sophisticated approaches and only now is the simplistic route being promoted. The problems these people fail to address are two fold. 1) Hydrobromination of saffrole is problematic. 2) The nucleophilic potential of an amine goes up with the degree of substitution. That means the reaction will not stop at the secondary amine MDMA but will aminate one or two more bromosafrole molecules to give a tertiary or quaternary amine and not the product you want. Reductive amination using sodium cyanoborohydride address this issue properly. Sodium borohydride is more accessible than sodium cyanoborohydride, but it will reduce the ketone to the secondary alcohol and so it is not used. Sodium cyanoborohydride used at neutral pH has the property that it will reduce the imine (Schiff base) formed between the ketone and methylamine much much faster that the imine. It is a selective reducing agent of the first rank. For research purposes the place to start for primary literature is the Merck Index. The entry for will lead you to the original literature which details the synthesis of sodium cyanoborohydride and also the typical procedures used in reductive amination or other selective reductions. Sodium cyanoborohydride can be made by reacting equimolar quantities of sodium borohydride and hydrogen cyanide in THF. One gram of hydrogen cyanide can kill one hundred men so do not attempt this unless you are a first rank chemist like me. The hydrogen cyanide is made following a procedure found in Organic Synthesis, Collective Edition. Work at the 50-100 gram scale in a distillation apparatus with ground glass joints. Emil Fischer established the practice where you must smoke cigarettes while you do this work. Nicotine accentuates the taste of HCN to give you an early warning of danger. In the absence of a fumehood wear scuba gear when disassembling the glassware. Store the liquid HCN frozen in the freezer. Combine equimolar quantities of NaBH4 and HCN in THF and allow the reaction to go to completion with the evolution of H2. Remove the THF with a rotary evaporator. If you do not own a rotary evaporator go out and buy one right now. The NaCNBH3 you made fresh is better than any you can buy. If you don't mind the heat, go out and buy the NaCNBH3 instead. More theory: the reductive amination of the ketone can lead to higher amines but this can be overcome with the use a five times molar excess of methylamine. That means that for this reaction you are going to want to make a large quantity of methylamine hydrochloride. 40% aqueous methylamine is easy to get and work with. Take equimolar quantities of 40% MeNH2 and concentrated HCl and make them as cold as you can in a deep freeze. Do the next part outdoors, clandestine and at night to avoid detection. Combine the two cold liquids quickly in a 1000 ml beaker. It will get real hot real quick and a lot of MeNH3CL smoke will be given off but you can put a cover on the beaker and the smoke will dissipate real quick so the neighbours will be unaware of what you did. Take the solution inside cool it and then remove all the water with your rotary evaporator to yield a good crop of white methylamine hydrochloride as a cake in your flask. So now you are all set to do your reductive amination of your ketone. You start with either phenylacetone or with 3,4MDP2P. Organic Synthesis, Collective Edition describes to methods for making the phenylacetone. One involves the condensation of acetic anhydride with benzylcyanide using sodium ethoxide, the other, the tube furnace method from phenylacetic acid. Or you made the 3,4MDP2P from isosafrole, 30% H2O2 and formic acid following the "Japanese Method" detailed somewhere in Chemical Abstracts. More theory: sodium cyanoborohydride is a selective reducing agent and it evolves hydrogen at a slow rate in solution. So you do not want to want to react at reflux. Reductive amination with sodium cyanoborohydride is best done in methanol at room temperature. The reaction is mostly over in a day and a half and 98% yield can be achieved in three days of waiting. Starting with a given molar quantity of ketone use a 25-50% molar excess of NaCNBH3 based on moles of hydrogen and a five times molar quantity of MeNH3CL in sufficient methanol to bring everything into solution. The beauty of this reaction is that you do not need expensive glassware of fixed capacity. You can work at humungous scale using even a plastic garbage can. After three days neutralize any unreacted NaCNBH3 using a calculated quantity of dilute HCl or acetic acid. Remove all of the methanol using your rotary evaporator. Then dissolve all of the cake in H2O. Add sufficient 5 N NaOH to cause the desired amine product to separate as a slightly brown layer floating on top of the aqueous layer. Here is where the work up can be slightly different depending on whether the product is methamphetamine or MDMA. The meth cook will sample the meth and continue his work in an energized manner for many more hours or days. Good meth cooks tend to be fastidious about the quality of the meth he makes and does. It is possible to just separate the top layer using a separatory funnel and to carry on. A better approach is to steam distill the amine out of the pot. The water in the aqueous layer is sufficient. The steam distillate is collected. Separation is enhanced by the addition of just sufficient 5 N NaOH. The floating amine layer is now clear. The top layer is removed with a separatory funnel an the bottom layer is washed a couple of times with a small quantity of ethyl ether which is combined with the top layer. You now have liquid meth with a small quantity of ether, a little dissolved water and perhaps some MeNH2 or unreacted ketone as a water white oily liquid. It is traditional to dry this over anhydrous NaSO4 or equivalent. Now set up your vacuum distillation apparatus with a water aspirator vacuum pump. If you do not own a distillation apparatus with ground glass joints go out and buy one right now. Depending on your vacuum the product will distill at 130 C or so and the liquid MDMA somewhat higher. With the pure liquid meth it is now time to create the hydrochloride. Many people think it is possible to create crystal meth using gaseous hydrogen chloride. I have never seen this to be true. What is obtained instead is an amorphous product. Why bother. Instead use your HCL gas generator to produce a quantity of anhydrous methanolic HCL in the case of MDMA or you can use aqueous HCL for meth. Combine equimolar quantities of HCl solution and amine product in methanol to neutrality. Use your rotary evaporator to remove solvent to yield a white cake in your flask. This is crude though pure methamphetamine HCl or MDMA HCL. It is now time to recrystallize your immensely valuable product. Dissolve the cake in the minimum quantity of hot isopropyl alcohol. Cool the IPA solution in the fridge. Crystallization may begin. Layer the solution on top with an equal quantity of anhydrous diethyl ether and put beaker into freezer overnight. I am told it is also possible to use methanol and MEK as solvent pairs. Overnight the ether layer will be seen to diffuse into the IPA and a massive crystallization has occurred. Stir if necessary. Some crystals will be needles upto one mm in length. You are laughing. Separate solvent from filter mass using a buchner filter and vacuum. Air dry for a while and then remove residual using a vacuum desiccator. You have a world class crystalline product. You have money in the bank. MDA by reductive amination with NaBH3CN 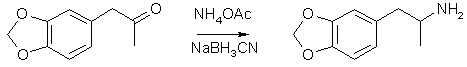
Into a 5 L flask on a stirplate place the following:
Note: add slowly and make sure that the acetate dissolves ok, the addition of acetate and methanol is endothermic and the solubility of it will go down, if you are not careful you will get a dome of acetate covering the bar and no movement of the solution!!! Into a 1L beaker place the following:
Stir with a glass rod, teflon stirbar retriever or something similar to knock out out all the chunks/goo of sodium cyano, note that it is fizzing and popping, do not reccomend breathing the fumes . Now into that 1 L beaker of fizzing cyano mix, add: 400g of MD-P2P. Stir it a little and quickly add it to the 5L flask. Oxygen degrades cyanoborohydride very quickly so this shouldnt take more than a minute or 2 from the time you open the container of cyano (pre weigh everything else.) Now take a balloon and fill it HALFWAY with nitrogen. Toys 'R' Us sells these punch balloons that fit nicely over a 24/40 joint. Anyhow take the N2 filled balloon and spray a little bit into the flask to get rid of the atmosphere in there and then place the ballon over the neck of the flask. Let stir 48-72 hours. Solution should be clear with a light tinge of whatever color your ketone was. Note: during the 2-3 day wait, make up about 2L of saturated NaOH (lye) solution and freeze it. After 48-72 hours, take the balloon off and distill off the methanol. It will be the first fraction that comes over and you should get what ya put in (~2 L). After the methanol is distilled off, cool the flask in an ice bath with stirring. If you got all of the methanol out whats left will more than likely polymerize (solidfy) into a big white mass. This is perfectly ok. Once the contents of the flask have cooled (<40C), CAREFULLY add 2L of 10% HCL(roughly 660ml muriatic and 1340ml distilled H2O.) Add slowly and make sure fizzing doesnt get out of hand or you will be wearing it!!! Let stir for a few minutes until all of the polymerization has broken up. Now dump half this into a 4 sep funnel and extract 2x200ml DCM. Then do the same for the other half. Save the dcm and freeze it, it contains any unreacted ketone and can be reused, simply boil off the dcm to know how much ketone ya got. Ketone can only be recycled once, after that it degrades. Now take the goods and put em back into that 5L flask in the ice bath (stirring). Now CAREFULLY add that cold saturated NaOH solution you made until you get to ~pH 11. It WILL fizz and you WILL paint your ceiling with this stuff if you dont add slowly and carefully. Don't add all 2L that you made up, just check the pH every now and then until it gets there. At about pH 9 you'll start to see amber oil fall out...thems the goods and its a damn pretty sight to see. Let it stir for a few minutes and then put what ya can in the sep funnel. Extract 3x200ml DCM AT LEAST...too much dcm dont hurt here. It usually turns red when ya do this too (anybody know why?) Anyhow that white cloudy water stuff will get clearer and clearer the more product you extract. Take the dcm/freebase oil and freeze till ya ready to xtallize. Dry thru a filter funnel of sodium sulfate and gas. Very clean and very pretty snow. You should get at least 200g of product. This method scales nicely and I dont know people who run this rxn at 3 times this scale in 12L flasks on a weekly basis. For you experienced bees, the 5x molar excess of acetate vs. the 10x doesnt seem to effect yields. Also while not having had the opportunity to test it, silica gel during that 2-3 wait has been said to improve yields ~25%. P.S. After adding the NaOH solution be sure that the entire contents of the flask have cooled <40C, otherwise when you add the DCM it will BOIL!!! |