Abstract
(±)-Lysergic acid (1) has been synthesized via an economical 8-step route from 4-bromoindole and isocinchomeronic acid without the need to protect the indole during the synthesis. Initial efforts to form the simpler 3-acylindole derivatives first and then cyclize these were unsuccessful in the cyclization step.
The ergot alkaloids pose an unusual opportunity for synthesis. The central alkaloids are all amides of lysergic acid (1) and all possess a broad range of pharmacological activity1-3. Only one of these, the diethylamide (LSD), however, is strongly and notoriously psychoactive. As such it is listed as a class I controlled substance. Since both the natural and synthetic derivatives are easily convertible to lysergic acid and so to its diethylamide, all of these are also controlled substances. As a result this potential pharmacological treasure is essentially unavailable for practical clinical testing.
Figure 1
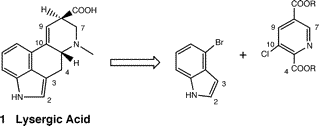
Bondset of lysergic acid synthesis.
We considered that a derivative of lysergic acid bearing an unremoveable substituent, like an added C-alkyl group, could not be converted to lysergic acid itself or its amide. Such derivatives would probably retain the broad pharmacological activity of the ergot family but might easily avoid the unique hallucinogenic property of LSD. This idea encouraged us to seek a short, practical synthesis route to lysergic acid suitable for incorporation of C-alkyl starting materials to create these derivatives.
Lysergic acid has already been synthesized about eight times4. The shortest path has 11 steps and none are serious candidates for practical manufacture. Every synthesis to date contains redundant protection/deprotection sequences, often as indole starting materials reduced and acylated, only at the end reconstituted to the indole form. To eliminate this redundancy we decided that the indole should be carried through intact.
Scheme 1
Attempt via Bondset ba Approachaa
Reagents: (i) EMgBr/ZnCl2/Et2O.
The simplest convergent bondset for assembling the lysergic skeleton should be the boldface bonds in ring C,
which just arise from indole and nicotinic acid starting materials. No previous syntheses had utilized this approach
except the Julia route, which did not carry the indole moiety through unchanged.
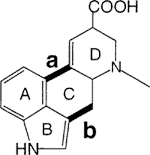
Of the two initial constructions necessary for the bondset in Figure 1 we began with the simplest (bond b) via an acylation of indole or 4-haloindoles5 2 with the acid chloride 3 from the commercially available 6-carboxynicotinic acid (isocinchomeronic acid), as outlined in Scheme 1. The acid was esterified, selectively hydrolyzed only at the 6-position6 with aqueous Cu(NO3)2, and converted (SOCl2) to 3. The Grignard reagents from 2 were acylated with 3 to form 4. However, a number of attempts at palladium-catalyzed cyclizations of 4 (X = Br or I) or reduced pyridine derivatives of 4 and their N-methyl salts were all unsuccessful.
We also considered that a thermal pericyclic cyclization of the anion 5 of 4 might be accessible with subsequent loss of HX to form 6, but several trials of this at elevated temperatures, with or without added base, led only to intractable tars.
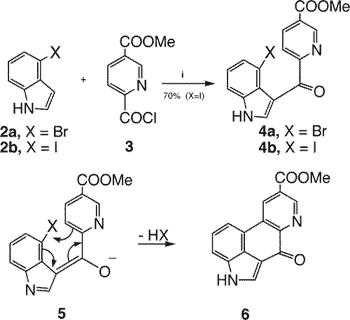
The alternative path to close ring C, making bond a first, was ultimately successful, as summarized in Scheme 2. For this approach we needed a nicotinic acid derivative with a halogen marking the 5-position.
Scheme 2
Synthesis of Lysergic Acid via Bondset ab Approachaa
Reagents: (i) Pd(PPh3)4/Na2CO3(aq)/EtOH; (ii) NaBH4,
CaCl2, EtOH; (iii) MnO2, CHCl3; (iv) NaOH, MeOH;
(v) NaBH4, TFA, CH2Cl2; (vi) MeI, CH2Cl2; (vii) NaBH4,
MeOH; (viii) NaOH, EtOH.
The common introduction of halogen on pyridines, via SOCl2 on the N-oxide7, provides only the ortho/para halides. However, sulfonyl halides can give rise to meta substitution8 and the reaction of the N-oxide of 6-carboxynicotinic acid with thionyl chloride affords9 the m-chloro derivative 7 on workup with methanol. We believe this results from first forming the normal, nonplanar p-chloro intermediate in Figure 2, which can then collapse via the pericyclic rearrangement shown and subsequent loss of the p-chloride to afford 7.
Figure 2
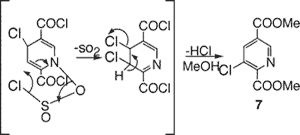
Proposed mechanism for formation of 7.
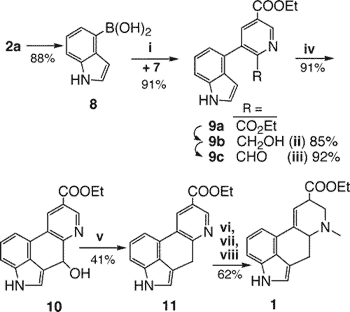
When the 4-haloindoles 2 were converted to the boronic acid 8 (via KH + BuLi and B(n-BuO)3), the Suzuki coupling was successful in forming 9a in 91% yield. While this work was in progress, a closely related reaction appeared in a note by Doll10 coupling 8 with 5-bromonicotinic ester, but the subsequent addition of the missing carbon 4 for lysergic acid failed.
We presumed that an appropriate base would easily initiate the cyclization of the diester 9a to the tetracyclic ketone corresponding to 10. However, treatment of the diester with NaH, even in glycol at 197°C, yielded only starting material. A number of attempts to cyclize the corresponding, very insoluble diacid with thionyl chloride or variants of PPA led only to recovery of diacid or intractable mixtures with no evidence of cyclization.
Following a similar but intermolecular version from Potier11 we reduced the ester selectively12 with NaBH4 and CaCl2 to 9b and then oxidized it to the aldehyde 9c with MnO2. The aldehyde cyclized at room temperature easily and quantitatively with only 2 mol% of NaOH to yield 10. Various efforts to cyclize the alcohol 9b or its tosylate to 11 all recovered only starting materials. The difference in the ease of cyclization of 9c over its precursors surprised us but we became convinced from models that there was severe steric resistance to the stereoelectronic demands for cyclization except for the aldehyde case.
Typical reduction13 of the indole-alcohol 10 with NaBH4/TFA afforded 11, which proved to be unstable, decomposing in a matter of hours. Accordingly, the remaining steps were carried through without isolation. Freshly prepared 11 was methylated directly with methyl iodide and the crude salt 12 reduced with excess NaBH4 in methanol to a mixture of methyl lysergate and its cis-isomer isolysergate in a 6:1 ratio, as a pale yellow solid.
These diastereomers are reported to be somewhat unstable14 and so were immediately hydrolyzed to lysergic acid with NaOH, which also equilibrated them to the more stable lysergic acid, which was then finally recrystallized to mp 241-242°C (lit. 242-243°C).
The 1H NMR spectra of the mixed esters were identical with spectra kindly provided by Prof. Ichiya Ninomiya, and the NMR spectra (1H and 13C) of the lysergic acid agreed with that of a natural sample kindly provided by Dr. David Nichols.
The last three operations (10 to 1) are carried out easily in good yield without isolation and purification; this result lends value to the initial conception in Figure 1 that the most economical synthesis of lysergic acid is one that originates in the two main starting materials, a simple indole and a nicotinic acid derivative, both retaining their aromaticity to the very end. This synthesis comprises eight steps from isocinchomeronic acid and 4-bromoindole and proceeds in an unoptimized overall yield of 10.6%. Chirality is only introduced in the final reduction step, and enantioselective measures for this reduction have not yet been examined, nor has the parallel synthesis of C-alkyl derivatives.
Experimental
1H and 13C NMR spectra were recorded on a Varian Unity Inova 400 MHz instrument at ambient temperature using TMS as internal standard and CDCl3 as solvent. Mass spectrometry was recorded on the Micromass QUATTRO II instrument. The solvents and reagents were purified by the following methods: diethyl ether, glyme and THF were distilled from sodium with benzophenone as an indicator. DMF, CH2Cl2 and xylene were distilled from calcium hydride. Benzene and toluene were distilled from P2O5. Methanol and ethanol were dried over magnesium. Triglyme was distilled from LiAlH4. Trimethylamine was distilled from NaOH. Anhydrous CaCl2 has been roasted in a crucible and allowed to cool in a desiccator.
1. 4-Bromo-indole (2a)
To a solution of 3-methoxycarbonylindole (7.0g, 40.0mmol) in TFA was added Thallium(III) trifluoroacetate (32.6g, 60.0 mmol) in TFA (140 ml), and the mixture was stirred for 2 hours at r.t. After TFA was evaporated in vacuo, a dark brown oil was obtained. This oil was dissolved in DMF (100 ml) and CuBr2 (35.8g, 160.0 mmol) was added. The reaction was stirred at 120°C for 1 hour then was cooled and CH2Cl2:MeOH (95:5, v/v) (300 ml) was added. Insoluble precipitates were filtered off through a plug of celite. The filtrate was washed with brine (100 ml x 2), and the organic layer was dried over Na2SO4. A crystalline material (7.60g) was obtained in 63% yield after the removal of solvent under reduced pressure. This material was directly subjected to decarboxylation for the preparation of 4-bromo-indole in the next step. To a solution of 4-bromo-indole-3-carboxylic acid methyl ester (5.06g, 20.0 mmol) in 200 ml methanol was added 200 ml of 40% aq. NaOH. The reaction was refluxed for 1.5 h with stirring. After evaporation of the solvent, the residue was poured into 200 ml water, and extracted with CH2Cl2:MeOH (95:5, v/v; 200 ml x 3). The extract was washed with brine, dried over Na2SO4, and evaporated in vacuo to leave a brown oil. Purification by chromatography using hexane:ethyl acetate (6:1) afforded a light colored oil (2.68 g) in 69.1% yield.
2. 4-Iodo-indole (2b)
To a solution of 3-methoxycarbonylindole (7.0g, 40.0mmol) in TFA was added Thallium(III) trifluoroacetate (32.6g, 60.0 mmol) in TFA (140 ml), and the mixture was stirred for 2 hours at r.t. After TFA was evaporated in vacuo, a dark brown oil was obtained. This oil was suspended in 450 ml H2O, and KI (19.9 g, 120.0 mmol) was added to this suspension. The reaction was stirred at r. t. for 2 hours. CH2Cl2:MeOH (95:5, v/v) (300 ml) was added to the reaction mixture and insoluble precipitates were filtered off through a plug of celite. The organic layer was separated and washed with aq. sodium thiosulfate then brine. Removal of the solvent left a brownish solid. Quick purification by a short plug of silica gel afforded a white solid (8.68g) as 4-iodo-indole-3-carboxylic acid methyl ester in 72.1% yield. This material was used directly in the next step. To a solution of 4-iodo-indole-3-carboxylic acid methyl ester (6.02g, 20.0 mmol) in 200 ml methanol was added 200 ml of 40% aq. NaOH. The reaction was refluxed for 1.5 h with stirring. After evaporation of the solvent, the residue was poured into 200 ml water, and extracted with CH2Cl2:MeOH (95:5, v/v; 200 ml x 3). The extract was washed with brine, dried over Na2SO4, and evaporated in vacuo to leave an off-white solid. Purification by chromatography using hexane : ethyl acetate (6:1) afforded a white crystalline solid (3.51g) in 72.3% yield.
3. Pyridine-2,5-dicarboxylic acid 5-methyl ester (3)
To a solution of pyridine-2,5-dicarboxylic acid dimethyl ester (5.91g, 30 mmol) in 100 ml methanol was added copper(II)nitrate trihydrate (14.5g, 60 mmol) in a 500 ml round bottom flask equipped with a reflux condenser and a stirring bar. The reaction was refluxed for 80 mins. A deep violet-blue precipitation was observed after 20 mins and lasted throughout the course of the reaction. The reaction was cooled to r.t., and the reaction mixture was reduced to 1/3 of its original volume. The deep violet-blue solid was collected by filtration and washed with cold methanol then cold water. This solid material was dissolved in 50 ml glyme, and H2S gas was bubbled into the solution. The black precipitate was formed in 2 mins, and the deep violet-blue solid disappeared after 15 mins. The black precipitate was filtered out through a plug of celite and the filtrate was concentrated to 20 ml. Excess hexanes were added into this solution, and a white solid was formed. The white solid was collected by filtration. Recrystallization of this solid from acetone afforded 4.83g product in 88.9% yield (mp 194.1-195.1°C, Lit6).
4. 6-(4-Iodo-indole-3-carbonyl)-nicotinic acid methyl ester (4b)
To a solution of EtMgBr (10 ml, 1.575 M in ether, 15.75 mmol) was added a solution of 4-iodo-1H-indole (3.65g, 15.0 mmol) in ether (anhydrous, 20 ml). The resulting twophase system was allowed to stand for 15 min under stirring whereupon ZnCl2 (15 ml, 1.0m in ether, 15.0 mmol) was added with stirring. The two-phase system was allowed to stand for 30 min when 6-chlorocarbonyl-nicotinic acid methyl ester (3.14g, 15.75 mmol) in anhydrous ether (10 ml) was added rapidly under vigorous stirring. The reaction mixture was allowed to stand for 2 hours whereupon NH4Cl (aq. sat. 25 ml) was added. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3x50 ml). The combined organic layer was washed with NaHCO3 (aq. sat. 25 ml) followed by brine (25 ml), and dried over Na2SO4. Removal of the solvent in vacuo afforded a yellowish solid. Recrystallization of this solid from acetone afforded 4.25g of the desired crystalline product (mp. 245.7-246.8°C) in 69.8% yield.
5. 3-Chloro-pyridine-2,5-dicarboxylic acid dimethyl ester (7)
Pyridine-2,5-dicarboxylic acid (8.35g, 50.0 mmol) was suspended in 250ml 0.2% (w/w) aq. Na2WO4 (0.5g) in a 1 liter round bottom flask. To this solution was added H2O2 in water (30% w/w, 8.5g, 75.0 mmol). The resulting mixture was stirred and heated at 80- 85°C for 10 hours. The resulting solid was collected by filtration and washed with cold water. Drying the material under vacuum overnight yielded 9.06 g product solid (mp. 253.2-254.1°C, dec; Lit.9 254°C, dec.) as pyridine-2,5-dicarboxylic acid N-oxide in 99% yield. This solid was used in the next step.
To a solution of thionyl chloride (9.52g, 5.84 ml, 80 mmol) in 200 ml CH2Cl2 was added DMF (2 ml) at 0°C. Pyridine-2,5-dicarboxylic acid N-oxide (3.66g, 20.0 mmol) was added into this mixture portionwise. The resulting mixture was heated at 65°C for 2 hours. The reaction was cooled to r.t. then placed in an ice-bath. The reaction was quenched with methanol (30 ml) slowly at 0°C. The solvent was removed in vacuo and the crude product was partitioned between CH2Cl2 (100 ml) and aq. NaHCO3 (sat. 50 ml). The organic layer was separated and the aqueous layer was extracted with additional CH2Cl2 (100 ml x 2). The combined organic layer was dried over Na2SO4 and the solvent was removed in vacuo. Purification of the crude oil through a short plug of silica gel using hexane : ethyl acetate (2:1) afforded a white solid (3.73g, mp. 126.0-127.0°C) in 81.2% yield.
6. Indole-4-boronic acid (8)
To a suspension of KH (4.57 g of a 30% suspension in mineral oil, 32.9 mmol) was added a solution of 4-bromo-indole (5.88g, 30.0 mmol) in anhydrous ether (25 ml). The reaction was stirred at r.t. for 30 min under N2 and the reaction was cooled in an acetone-dry ice bath (-78°C) with stirring. Precooled t-BuLi solution in hexane (33.0 ml, 66.0 mmol) was cannulated into the reaction and the reaction was kept stirring for another 20 min. Neat B(n-BuO)3 (24.1 ml, 90.0 mmol) was added into the reaction by syringe under vigorous stirring. The reaction mixture became thick when it was allowed to warm to r.t., and more anhydrous ether (30 ml) was added under N2. The reaction was allowed to stand overnight at r.t. with vigorous stirring. The thick reaction mixture was diluted with more anhydrous ether and then transferred slowly into 1 M aqueous H3PO4 (300 ml) at 0°C. The mixture was stirred at r.t. for 40 min, and extracted with ether (100 ml x 30). The combined organic layer was extracted with 1 N NaOH (50 ml x 3). Ether (100 ml) was added to this aqueous solution and the mixture was acidified to pH = 2 using 1M H3PO4. The organic layer was separated and the aqueous layer was extracted with ether (100 ml x 2). The combined ether layer was dried over Na2SO4 and evaporation of solvent in vacuo left a beige solid (4.24g, 88.0%).
7. 3-(4-indolyl)-pyridine-2,5-dicarboxylic acid diethyl ester (9a)
Into 500 ml anhydrous toluene in a 1 liter round bottom flask equipped with stirring bar was bubbled in a stream of argon via a needle for 30 min. Pd(PPh3)4 (0.878g, 0.75 mmol) and 3-chloro-pyridine-2,5-dicarboxylic acid dimethyl ester (3.44g, 15.0 mmol) were added into this solvent and the resulting mixture was stirred at r.t. under argon for 1 hour. A solution of indole-4-boronic acid (1.86g, 11.5 mmol) in 50 ml EtOH and a solution of 2M aqueous Na2CO3 (11.5 ml) was added into the reaction mixture at r.t. under argon. The mixture was heated under argon with vigorous stirring at 105°C for 8 hours. The reaction mixture was cooled and brine (200 ml) was added. The organic layer was separated and aqueous layer extracted with additional CH2Cl2 (2x100 ml). The combined organic layer was dried over Na2SO4 and evaporation of the solvent left a yellowish solid. Purification of the crude material through a short plug of silica gel (hexane:ethyl acetate, 1:2) afforded a yellow solid. The TLC of this material showed it to be a mixture of three different compounds due to ester exchange. This solid was dissolved in 500 ml EtOH and the solution was stirred overnight at r.t. in the presence of cat. HCl in diethyl ether. A single compound (3.54g) was obtained (mp. 212.3-213.0°C) in 91.0% yield.
8. 6-Hydroxymethyl-5-(4-indolyl)-nicotinic acid ethyl ester (9b)
To a solution of 3-(4-indolyl)-pyridine-2,5-dicarboxylic acid diethyl ester (0.34g, 1.0 mmol) in anhydrous EtOH (10 ml) was added Ba(BH4)2 (24.6mg, 0.65 mmol) followed by CaCl2 (44.3mg, 0.4 mmol) at 0°C. The reaction was warmed to r.t. and stirred for 2 hours. 1 M H2SO4 (1 ml) was added to the reaction, and the resulting white precipitation (CaSO4) was filtered out through a plug of celite. The filtrate was concentrated and partitioned between ethyl acetate (20 ml) and NaHCO3 (aq. 15 ml). The organic layer was separated and the aqueous layer was extracted with additional ethyl acetate (2x20 ml). The combined organic layer was dried over Na2SO4, and the solvent was removed. The crude material was purified by silica gel chromatography using hexane-ethyl acetate (3:1 to 1:1). A colorless solid (mp. 198.1-199.0°C) was obtained (231 mg) in 78% yield.
Alternatively, the product could also be made using Ca(BH4)2 as the reducing reagent in the same solvent and temperature for same period of time. The NMR, mass spectrum, elemental analysis and mp. of this product were identical to that of the product obtained by the previous method. The yield of this reaction was 85%.
9. 6-formyl-5-(4-indolyl)-nicotinic acid ethyl ester (9c)
To a solution of 6-hydroxymethyl-5-(4-indolyl)-nicotinic acid ethyl ester (296 mg, 1.0 mmol) in 5 ml CH2Cl2 was added freshly made MnO2 (870 mg, 10.0 mmol). The reaction was stirred at r.t. for 2 hours then filtered from the solution through a plug of celite, and the solvent was removed in vacuo. Purification of the crude material by silica gel chromatography using hexane-ethyl acetate (2:1) afforded a yellow solid (271mg, 92%) with mp. 198.4-199.2°C.
10. 6-Hydroxy-4,6-dihydro-indolo[4,3-fg]quinoline-9-carboxylic acid methyl ester (10)
To a solution of 6-formyl-5-(4-indolyl)-nicotinic acid ethyl ester (117.8 mg, 0.4 mmol) in anhydrous methanol (1.0 ml) in a 2 ml conical vial was added 0.5 M NaOMe/MeOH ( 16.0 µl, 0.008 mmol). The reaction was stirred at r.t. for 2 hours. Solid precipitated out of the solution during the course of the reaction, and the starting material disappeared completely after 2 hours as indicated by TLC. The solution was cooled to 0°C and the liquid was removed with a pipette. The remaining solid was recrystallized from MeOH to afford 102.0 mg yellow crystalline solid (mp. 234.6-235.8°C) in 91% yield.
11. 4,6-Dihydro-indolo[4,3-fg]quinoline-9-carboxylic acid methyl ester (11)
To a solution of 6-hydroxy-4,6-dihydro-indolo[4,3-fg]quinoline-9-carboxylic methyl ester (90 mg, 0.32mmol) in 10 ml anhydrous THF was added BH3 in THF (1.0 M, 0.64 ml, 0.64mol) under argon. The resulting mixture was stirred at r.t. for 2 hours. The TLC showed the disappearance of the starting material and a new fluorescent spot under UV on TLC. The solvent was removed in vacuo and the crude material was partitioned between CH2Cl2 (3 ml) and aq. NaHCO3 (sat., 2 ml). The organic layer was separated and the aqueous was extracted with additional CH2Cl2 (2x3 ml). The combined organic layers were dried over Na2SO4 and concentrated. The crude material was purified by silica gel PTLC using CH2Cl2:MeOH (98:2). A white solid was obtained (mp. 212.9-213.9°C, 35.2 mg) in 41% yield.
12. (±)-Lysergic acid (1)
To a solution of 4,6-dihydro-indolo[4,3-fg]quinoline-9-carboxylic acid methyl ester (29.2 mg, 0.116 mmol) in CH2Cl2 (1ml) was added MeI (33.0 mg, 14.6 µl, 0.24 mmol) at 0°C. The reaction was stirred at 0°C for 2 hours and the starting material disappeared after 2 hours as indicated by TLC. The solvent was removed in vacuo and the crude product was dissolved in methanol (1 ml). To that mixture was added NaBH4 (15.2 mg, 0.4 mmol) and the reaction was stirred at r.t. for 5 min. The organic solvent was removed in vacuo and the remaining solution was partitioned between CH2Cl2 (2 ml) and water (2 ml). The organic layer was separated and the aqueous layer was extracted with additional CH2Cl2 (3 ml x 2). The organic layers were combined and dried over Na2SO4. The solvent was removed and the crude material was purified by PTLC (CH2Cl2:MeOH, 98:2). A white solid was obtained (21.4 mg, 65%). 1H NMR showed it to be a mixture of methyl lysergate and methyl isolysergate in 6:1 ratio using N-methyl as the integration indicator.
To a solution of methyl lysergate & methyl isolysergate (6:1 mixture above, 15.6 mg, 0.028 mmol) in ethanol (0.5 ml) was added 1 N NaOH (0.5 ml). The reaction was heated at 35°C for two hours. 0.1 N HCl solution was used to carefully adjust the pH to 6 and the solid material was collected by removing the liquid. The solid was recrystallized from ethanol to afford 12.2 mg, (95%) of lysergic acid.