Quantitative Conversion of Codeine to HydrocodoneSynthetic Communications 30(17), 3195-3201 (2000)IntroductionIn the course of a project involving the synthesis of potentially very specific opiate antagonists, we recently required multi-gram quantities of hydrocodone as the free base. This material is commercially available only as the bitartrate salt, and the price is prohibitively expensive for a starting material (ca $450/g), especially considering that the tartaric acid would immediately be removed in the planned sequence. Consultation of the literature revealed several syntheses of hydrocodone, usually beginning with either codeine or thebaine. The most promising of these appeared to be a method published by Rapoport et al [J. Org. Chem. 15, 1103 (1950)], which entailed the catalytic hydrogenation of codeine in dilute acetic acid followed by an Oppenauer oxidation, providing a reported yield of 83% of the desired product. Using this sequence as a starting point, we now report a very facile sequence that provides pure hydrocodone in nearly quantitative yield from codeine, which can be obtained for $17/g. In our hands, the catalytic hydrogenation of codeine in dilute acetic acid to dihydrocodeine gave material that was rather gummy and not suitable for carrying on the oxidation step. We determined after considerable experimentation that ethyl acetate constituted an ideal solvent. Hydrogenation of codeine at room temp in a Parr apparatus at 35 psi hydrogen for 2-3h afforded a quantitative yield of pure dihydrocodeine, with a melting point higher than any reported value. Considerable time was invested in the oxidation step, but the notorious acid-lability of the morphinan skeleton precluded the utilization of any of the alternative methods tested, therefore we focused our attention on streamlining or refining the reported Oppenauer method. In Rapoport's paper, potassium tert-butoxide was prepared from potassium and tert-butyl alcohol, but instead we utilized a comercially available 1M solution of potassium tert-butoxide in THF, whioch worked very well as long as the THF was completely removed prior to the addition of the oxidation reagents. Also, attempted substitution of toluene for benzene proved unsatisfactorily, only unreacted starting material was recovered. Once the solution of potassium tert-butoxide in benzene was prepared, a solution of codeine and benzophenone was added, and the solution refluxed for a short time, whereupon a standard extraction provided pure hydrocodone in nearly quantitative (99%) yield. 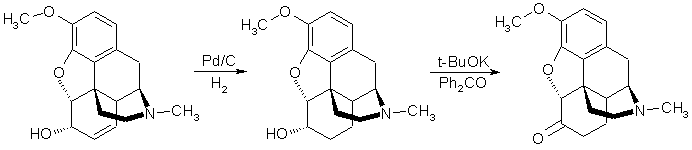 Dihydrocodeine from CodeineIn a Parr hydrogenator jar, codeine (2.0g, 6.7 mmol) and 10% Pd/C (200mg) were combined in ethyl acetate (75ml). This mixture was hydrogenated at room temp at 37 psi for 2h. During this time, the hydrogen pressure dectreased to 34 psi within 30 min, was increased back to 37 psi, and thereafter remained constant for the duration of the reaction. The mixture was filtered through Celite, the filter cake washed with 2x10ml ethyl acetate, the resulting solution stripped of solvent using a rotary evaporator by high vacuum. The product foamed considerably upon initial application of high vacuum, so care was taken not to lose any product. Leaving the product on high vacuum overnight afforded a white, crunchy, crystalline solid (2.0g (99.3%), mp 113-4°C [lit. 112-3°C]). Hydrocodone from DihydrocodeineA 100ml round-bottomed flask was weighed and equipped with a vigreaux column and distillation head along with benzene (20ml). Using a syringe, potassium tert-butoxide (5ml of a 1.0M solution in THF, 5mmol) was added to the flask and the resulting yellow mixture was distilled under a dry nitrogen atmosphere. Fresh benzene was added periodically until the head temperature reached 80°C and remained constant. When about 20ml of benzene had been distilled, the apparatus was allowed to cool and the reaction flask was set up for reflux. In a 25ml round-bottomed flask was prepared a solution of dihydrocodeine (500mg, 1.66 mmol), benzophenone (3g, 16.5 mmol) and benzene (15ml), which was added in a single portion via syringe to the reaction flask, accompanied by a slight exotherm. The reaction flask was flushed with nitrogen and was then gently refluxed for 2.5h. During the reflux, the formation of a greenish-brown precipitate was observed. The reaction mixture turned a deep green color by the end of the reaction time and contained large quantities of a white solid.At this time, the reaction flask was cooled in an ice bath and hydrochloric acid (15ml of a 3M solution, 45 mmol) was added to the flask. The resulting mixture was transferred to a separatory funnel and the layers were separated. The organic layer was extracted twice with hydrochloric acid (15ml of a 3M solution, 45mmol). The first aqueous layer and the extracts were combined and washed with 2x15ml diethyl ether. The ether layers were discarded. The aqueous layers were then basified with 20% aqueous sodium hydroxide solution, causing the separation of a white clumpy precipitate. This mixture was extracted three times with 100ml ethyl acetate. The combined organic extracts were dried with magnesium sulfate, stripped of solvent on a rotary evaporator, and placed on high vacuum to remove residual traces of solvent. This protocol afforded 492mg (99%) of a snowy-white solid, mp 195-6°C (lit. 194-5°C). |