Best Method for Hydrochloric Acid Gasby Argox[ Back to the Chemistry Archive ] Background: The experienced chemists on this forum will probably scoff at the notion that anything so simple as hydrochloric acid gas (hydrogen chloride or HCl(g)) would require careful explanation as to proper methods, set-up and precautions. The grim reality is that novice chemists have killed themselves as a result of ignorance regarding HCl(g). A cursory review of Rhodium’s page and the Hive search engine (TFSE) regarding methods to produce HCl(g) and store it for future use reveal incomplete information, poorly written information, and a certain degree of debate among Hive members as to the best method. This document provides outstanding examples of ghetto devices for the manufacture of HCl(g), most of them dangerous, none reusable, and all obvious evidence of illicit activity. HCl(g) is not illegal to own or make. It is sold commercially as a compressed gas at 613 psi in CS cylinders. The commercial cylinder requires a CGA 330 fitting and single stage SS regulator (USD 350). The regulator must be purged with nitrogen after every use; even so it has a limited life span and will require refit or replacement on a regular basis. Commercial cylinders of HCl are regulated. The supplier is required to fill out certain forms and send the data to certain government agencies with every purchase. Most chemists, legitimate and clandestine, prepare HCl(g) in the laboratory, to avoid the cost, hassle, and danger of compressed gas. A review was conducted of four OTC methods, and the best method was determined. Methods: Four common OTC methods were examined:
All four methods produce HCl(g) in varying yields. However, one method was clearly superior to the others based on yield, speed of reaction, and, most importantly, in situ clean-up and reload of reactor. The best method to produce HCl(g) according to these criteria is reaction 1 - dripping muriatic acid over sulfuric acid. For the benefit of the novice, a detailed discussion of this method follows. In addition, two commonly used solvents were examined (acetone and IPA) to determine which was the best for collecting and storing HCl(g), since there are empirical benefits to be obtained by collecting and storing HCl(g) for future use in order to precisely dose certain bases, rather than the more uncertain, unwieldy and dangerous practice of direct gassing those bases. It was determined that IPA is superior to acetone. Discussion: Best Method. Dripping muriatic acid over sulphuric acid. HCl(aq) + H2SO4 => HCl(g) + H2SO4(aq) This method is superior to others for the following reasons:
Obtaining ingredients. To produce HCl(g) with this method in North America one should buy muriatic acid and drain cleaner from the (big orange) hardware store and 99% isopropanol (IPA) from any drug store. The novice should make sure the label of the muriatic acid says "contains hydrochloric acid" and "muriatic acid commercial 31%," make sure the label of the drain cleaner says "contains sulfuric acid," and make sure the label on the IPA says 99% and "isopropyl alcohol." Testing ingredients. There is no need to test the muriatic acid, other than making sure that the label does indeed say muriatic acid. Confirmation that it is muriatic acid comes when the top is taken off the bottle and the characteristic fog of acid vapor rises out of the bottle. Do not breath this vapor: it is pure hydrochloric acid produced by the reaction of HCl(g) with water vapor in the air. Test the drain cleaner/sulfuric acid by weighing 500 cc. Drain cleaner/sulfuric acid is a heavy dark liquid and 500 cc should weigh between 917 and 920 grams. If 500 cc weighs substantially less than 917g, then it has been diluted with water and cannot be used. Look for a different brand of drain cleaner or obtain commercial-grade sulfuric acid, a non-watched chemical. The specific gravity of sulfuric acid is 1.84. 1000cc weighs 1840g. 500cc weighs 920g. Test 99% IPA by dripping one drop onto a clean mirror and holding the mirror up against a light bulb. If the IPA is truly 99%, then the drop will evaporate without leaving any smaller drops on the mirror. The presence of a number of miniscule cloudy drops on the mirror after the initial drop has evaporated indicates water contamination. If this be the case, buy another brand of IPA or dehydrate the IPA with 3A molecular sieve, anhydrous MgSO4 or similar anhydrous salt. The various methods of removing moisture from alcohols and other solvents have been thoroughly discussed in numerous posts. Setting up equipment This discussion of equipment is predicated on two personal philosophies:
The novice may find this discussion of laboratory glassware daunting. However, the benefits obtained from using proper glassware outweigh the costs, as per the above two points. (Note: the Rhodium chemistry page contains a write-up by Psychokitty describing a gas generator using beer bottles and syringes. That write-up is a "ghetto" alternative for the novice CC who needs only grams of HCl(g) rather than kilos and who cannot afford good equipment.) The gas generator consists of four main pieces:
These are connected by a claisen adapter, two 3-way vacuum adapters (suggested is a custom vacuum adapter that includes a stopcock for pressure relief), and joint-to-hose adapters. A stopcock or way of relieving line pressure should be placed between the reactor and the bubbler, and the bubbler and the receiver. All glassware should use standard taper joints, 24/40 is fine (it is suggested that rubber stoppers not be substituted for standard taper joints, rubber stoppers will degrade in the presence of HCl and eventually leak). Lab-grade silicone grease must be used on all glass joints (vaseline should not be substituted for lab-grade silicone grease). Also suggested is that each glass joint be secured with a Keck clip (24/40 Keck clips are green) or SS wire clip. All of the above glassware should probably be part of any novice chemist’s equipment inventory. In addition, to properly perform this method for producing HCl(g), the chemist should also purchase 2 magnetic stirrer hotplate combos and several magnetic stirbars (egg-shaped stirbars must be used for RB flasks). These stirrer/hotplates will be useful for many other reactions and will form an essential part of the equipment for other syntheses. None of this glassware or equipment is watched or controlled or illegal to own, as long as any RB flask is not 10L, 12L, 20L or 22L in size. A 2L equalizing addition funnel, a 5L FB or RB flask for the reactor, a 500 ml RB flask for the bubbler, and a 1L RB or ehrlenmeyer flask for the receiver are sufficient to produce substantial - even industrial - quantities of HCl(g) in a day, plus this glassware can be used for many other interesting reactions. 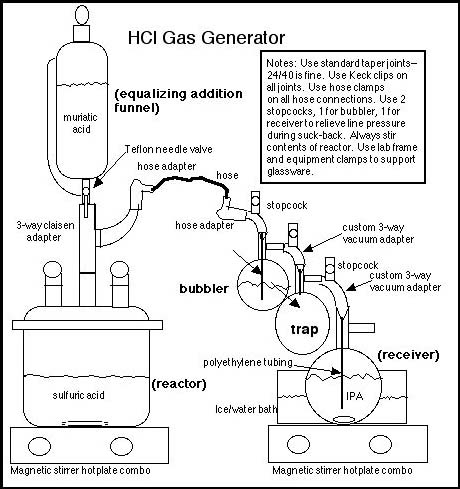
Theory. Muriatic acid, a mixture of water and HCl, is slowly dripped into a stirred flask containing sulfuric acid. Sulfuric acid has a strong affinity for water. The sulfuric acid dehydrates the muriatic acid, absorbing water and liberating HCl(g). The liberated HCl is then bubbled through a smaller quantity of sulfuric acid to remove any trace of water vapor, and then the dry gas is absorbed in stirred, cold IPA. By weighing the IPA before and after gas absorption, the exact production of HCl can be determined. This exact amount of HCl is very useful in subsequent reactions, since stoichiometric amounts of HCl can be dosed into base/solvent mixtures, in contrast to direct gassing of such mixtures, which is a "hit or miss" and exothermic operation. The HCl/IPA mix is stable and can be stored for future use, unlike HCl/acetone mix, which quickly degrades. IPA is not hygroscopic as is acetone, another important consideration. (Bees may note that this information is in contrast to several major Hive write-ups where HCl loading into acetone and/or direct gassing is recommended.) Practical application. Use 2 parts by volume of sulfuric acid/drain cleaner for every 1 part of muriatic acid. Put another way: use 2 bottles of drain cleaner for every 1 bottle of muriatic acid. (This will provide an excess of sulfuric acid necessary for proper dehydration of the muriatic.) The most important consideration is to constantly stir the sulfuric acid/drain cleaner while muriatic acid is being dripped. If the sulfuric acid is not stirred, a layer of muriatic acid will form on top of the sulfuric acid and when the flask is moved for any reason the two layers will mix with violent release of HCl(g). THEREFORE always stir the sulfuric acid while adding muriatic acid, this stirring cannot be avoided without disasterous results--so the magnetic stirrer is essential. Repeating again, ALWAYS stir the sulfuric acid while adding muriatic -do not shake by hand- stir with a magnetic stirrer and telflon coated stirbar. The gas bubbler is another important part of the set-up because it allows visual evidence of how much gas is being produced and it removes any water vapor that might come over with the HCl(g). The goal is to produce anhydrous HCl(g), since water vapor is detrimental to most hydrochloride crystallizations for which one would use HCl/IPA. Since HCl(g) is completely absorbed by cold IPA to a loading of 25% or greater, there will be no bubbles coming out of the receiver flask after the initial air is driven out of the gas generator set-up, therefore the bubbler provides the only visual indication as to whether the gas generator is going full blast or has stopped or has sprung a leak (although in the case of a leak, the characteristic mist of HCl(g) and the smell will be evident). Suck-back. Suck-back occurs when the amount of gas being generated cannot overcome the rate of absorption. A trap must be placed between the gas generator and the receiver as a safety precaution to collect any IPA sucked-back. To control suck-back, a stopcock placed before the bubbler and before the receiver will allow the operator to relieve line pressure. Additional tips. Do not use a dispersion tube while absorbing HCl(g) into IPA, as has been recommended in other posts. The dispersion tube is not needed since HCl(g) is immediately soluble in cold IPA. The dispersion tube could increase line pressure and cause a joint or flask to pop. 3/8" ID polyethylene tubing such as can be purchased at the (big orange) hardware store is sufficient for both the bubbler and the receiver. Stainless steel tubing is not recommend -this is in contrast to what has been suggested in another major Hive write-up- since HCl will corrode stainless steel and chromium/nickel/iron chlorides will go into solution and possibly contaminate the final product. Telfon hose is recommended for glass joint to hose connections; however, the clear vinyl hose found at the (big orange) hardware store can be used once. All hose connections must be fastened with hose clamps (obtained from any hardware store). With proper fittings this apparatus will not leak and will not smell, but working under a fume hood is always a good general practice. Yield. Yield is close to theoretical. Over 300g of HCl(g) will be produced from a 900ml bottle of muriatic acid in about 2 hours of slow dripping. Once the gas production begins to diminish, the sulfuric acid in the reactor can be heated and additional HCl(g) recovered (this is where the stirrer-hotplate combo comes in handy). If more than 10kg of HCl is needed in one day, then acid addition will have to be much more rapid, requiring larger size flasks and larger bore tubing to accomodate the increased reaction rate, but the novice should not need more than a few kilos of gas in one day, which can be generated with a 5L reactor. Clean-up and reloading. The most important advantage of this method is the fact that without taking the apparatus apart, the spent sulfuric acid can be pumped or siphoned (by vacuum) out of the reactor and then poured into a plastic pail of cold water and flushed down the toilet (after all, the label did say "drain cleaner"). Local water treatment plants can handle sulfuric acid no problem and those drains sure get a good cleaning. Reload is as simple as pouring or pumping another couple of bottles of drain cleaner into the reactor. None of the other common methods are as simple to unload and reload, their yields do not come close to theoretical, they are not appropriate for larger scale production. A discussion of the other methods follows: Other methods: Reacting sulfuric acid and table salt. Note that contrary to many posts and even government web pages this reaction produces sodium bi-sulfate, not sodium sulfate. Therefore the sulfuric acid:salt mole/mole ratio is 1:1, not 1:2. The reaction mechanism is: H2SO4 + NaCl => HCl(g) + NaHSO4 (The reaction of sodium bi-sulfate and table salt to produce hydrogen chloride only takes place at elevated temperature (+250ºC).) Hive Bees fervently defend this method of dripping sulfuric acid over table salt, yet it is inferior to the recommended method for these reasons:
A third method is heating table salt and sodium bisulphate. The reaction mechanism is: NaCl(s) + NaHSO4(s) + delta temp. => HCl(g) + Na2SO4(s) This method is also inferior for the following reasons:
A fourth method is boiling muriatic acid. The idea is to reduce the solubility product of HCl in water by boiling muriatic acid. However, hydrogen chloride forms a constant boiling mixture with water, such that a great deal of water vapor comes over with the HCl and quickly dilutes the sulfuric acid bubbler. Yield is unacceptable compared to any of the above reactions. Although the reactor can be pumped or siphoned out and reloaded, the remaining liquid still contains substantial amounts of HCl which even cooled and poured into water releases HCl vapor into the work area, very unpleasant to handle, and not something to pour down the drain since it is highly corrosive. An inferior method and not recommended at all. Summary: Of the four common methods to produce HCl(g) using OTC ingredients, one method was shown to be superior in terms of yield, reaction time, controllability, and ease of unload and reload, which is the most important aspect for larger-scale production. The superior method is dripping muriatic acid into sulphuric acid, preferably with the gas generator equipment described. It was also determined that IPA is superior to acetone as a solvent in which to store HCl for future use, since it will not degrade over time, is non-hygroscopic, and is compatible with common processes that require hydrogen chloride to precipitate hydrochloride salts from solvent/base mixes. It is a personal opinion that HCl(g) should be dissolved into cold IPA regardless of whether gas is generated as described above or obtained from a compressed gas cylinder, before dosing a solvent/base mix for two reasons:
|