(S)-fenfluramineby hypo(format & minor edits by metanoid) [ Back to the Chemistry Archive ] Bull. Soc. Chim Fr. (1993) 130, 450-458 BP 118 76134 Mont-Saint-Aignan Cedex, France Adresse actuelle: Institut de Recherches Servier, 11, rue des Moulineaux, 92150 Suresnes, France Société ORIL, 13 rue Auguste Desgenétass, 76210 Bolbec, France (reçu le 30 octobre 1992, accepté le 28 mars 1993) Summary - Syntheses of (S)-fenfluramine from (R) or (S)-1-[3-(trifluoromethyl)phenyl]propan-2-ol. (R) and (S)--1-[3- (trifluoromethyl)phenyl]propan-2-ol 3 are useful intermediates in the synthesis of fenfluramine (S)-1. They can be obtained from optically active propylene oxide (R) or (S)-7. The alcohol (R)-3 was transformed in two steps into (S)-fenfluramine using the action of ethylamine on a sulfonate (R)-4. We describe a new one-pot synthesis for (S)-fenfluramine from the azide (S)-5, which was obtained from the alcohol (R)-4 in two steps. We also propose an original and rapid procedure to transform the alcohol (S)-3 into (S)- fenfluramine via the chloride. (R)-14 and the azide (S)-5, without preliminary inversion of the alcohol. All of these reactions have been achieved without any loss of chirality. 1-[3-(trifluoromethyl)phenyl]propan-2-ol / fenfluramine / asymmetric synthesis / chiral methyloxirane / nucleophilic substitution Introduction Fenfluramine 1 is the active ingredient of a obesity drug acting on the digestion of carbohydrates, the activity being restricted mainly to the S enantiomer [1, 2], which can be obtained by separation of the diastereoisomers [3] or by preferential crystallisation of derivates, which were identified of being conglomerates [4]. Only two syntheses of optical active fenfluramine have been described until now: one by stereoselective reduction of the imine derived from the ketone 2 and (R) or (S)-alpha-phenylethylamine [5], the other starting from (S)-alanine [6]. Two recent publications [7, 8] about the synthesis of (S)-fenfluramine via the intermediate alcohol (S)-3 (scheme 1) made us publish our previous results [9]. Through yeast reduction of the ketone 2, the authors obtain the alcohol (S)-3, the configuration of which they inverse in three steps. The alcohol (R)-3, via the intermediate tosylate (R)-4a and further the azide (S)-5 leads to the amine (S)-6 after reduction and finally to the (S)-fenfluramine (S)-1 after reductive amination in presence of acetaldehyde: 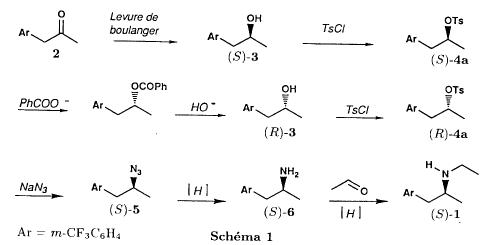 The (S)-fenfluramine is such obtained in 7 steps starting from the alcohol (S)-3 or in 4 steps from the alcohol (R)-3. In this article we present a new way of preparing the two enantiomers of the alcohol 3, a new two step synthesis of (S)-fenfluramine starting from the alcohol (R)-3, a one step synthesis of (S)-fenfluramine starting from the azide (S)-5, which doesn't pass over the intermediate primary amine (S)-6 and finally a much faster process (3 steps) of preparing (S)-fenfluramine starting from the alcohol (S)-3. Results and discussion Synthesis of 1-[3-(trifluoromethyl)phenyl]propan-2-ol (R)-3 The racemic alcohol is seldom mentioned. It's one of the metabolites of fenfluramine in the human body, secreted in urine [10]. It can also be obtained by metabolic transformations of an oxime by different kinds of microorganisms [11]. It was used as an intermediate for the synthesis of a family of anorexics [12] and a family of antispasmodic and psychotherapeutic agents [13]. It was obtained by the reaction of methyloxirane 7 with the magnesium compound 8, with a yield of 50%. This same reaction was described earlier as being little regioselective [14], a fact we observed too [9]. The only synthesis of the optically active alcohol 3 is the reduction of the ketone 2 with yeast as described above. One abtains the S enantiomer, the R enantiomer is obtained by inversion. On our part we used the condensation of the commercial [15] methyloxirane (R)-7, of which many syntheses are known [16], with the magnesium compound 8 and cuprous chloride [17, 18]. 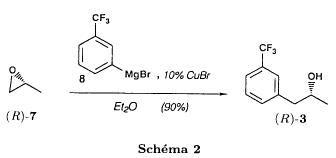 The yield is about 90% and the reaction very selective (purity GC: 93%). The optical purity of the methyloxirane 7 was determined by 1H-NMR in presence of the europium complex Eu(hfc)3 [19]. The optical purity of the alcohol (R)-3 was obtained by 1H-NMR and HPLC over silica of the Mosher derivate [20]. The comparison of these values show that the chiral centre is preserved. This procedure has the advantage of allowing us the preparation of the alcohol (S)-3 with the same reaction, because the methyloxirane (S)-7 is also commercially available and multiple syntheses are known [21]. Two step synthesis of the fenfluramine (S)-1 starting from the alcohol (R)-3 With the goal of obtaining the simplest procedure we have studied at first the transformation of the alcohol (R)-3 into fenfluramine (S)- 1 in two steps via the intermediate of the easily obtained sulfonates (R)-4: 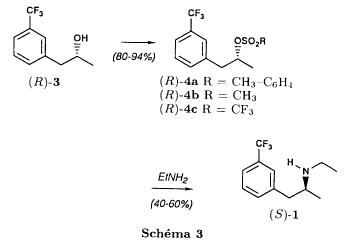 The substitution of the mesitylate (R)-4b and the tosylate (R)-4a with ethylamine was realised with medium yields always between 40 and 50% in spite of the large number of conditions tested: solvents (DMSO, DMF, ethanol, ethylamine), different dilutions (in proportions from 1 to 5) and temparatures from 50 to 160°C (with different times of contact). With the triflate (R)-4c the yield of the substitution is 60% but under non comparable conditions (-20°C in acetonitrile) because of its higher reactivity. In all cases the non aminated, and thus easily separated, byproducts are mainly the alkenes 9, 10Z and 10E (10E >> 10Z > 9). 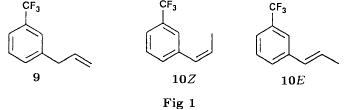 The enantiomeric purity of the amine (S)-1 is analysed by HPLC chromatography through silica of the camphanylated derivate [22] and compared to the previously analysed alcohol (R)-3: we have thus shown that the optical centre is conserved during the nucleophilic substitution. One had indeed to fear that due to the participation of the aromatic ring as neighbour group there could be partial or complete racemisation with an phenonium ion as intermediate. With the results obtained, which match with the literature [23-26], one can suppose that the trifluoromethyl group in meta position is sufficiently deactivating the aromatic ring in order to prevent participation in the substitution. We probably have thus in our case a pure nucleophilic SN2 substitution in competition with an elimination reaction. We believe that this elimination reaction is due to the simultaneous nucleophilic and basic properties of the ethylamine. Although the yields are medium, this method has the advantage of being relatively fast because it permits to prepare fenfluramine (S)-1 starting from the alcohol (R)-2 in two steps instead of four [7, 8]. As far as we know it was never mentioned in literature. Synthesis of fenfluramine (S)-1 from 2-azido-1-[3-(trifluoromethyl)phenyl]propane (S)-5 The substitution of the mesylate (R)-4b by sodium azide (scheme 4), an only slightly basic nucleophile compared to ethylamine, forms no elimination side products. One obtains the optically pure azide (S)-5 with a yield of 95%. The enantiomeric purity couldn't be directly analysed on the azide (S)-5. Only for analytical purposes did we reduce it into the amine (S)- 6. Among the numerous methods for the reductions of azides to amines mentioned in the literature [27] we chose the catalytic hydrogenation with 5% Pd on calcium carbonate at standard temperature and pressure [27f]. The HPLC analysis through silica column of the champhanyl derivate [22] of the amine (S)-6 such obtained shows that the enantiomeric centre was totally inverted during the substitution when compared to the enantiomeric purity of the alcohol (R)-3. 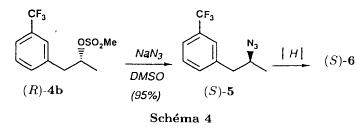 The reductive amination of the amine 6 in presence of acetaldehyde is known for a long time [28]. It was used recently in the works listed in the introduction [7, 8]. On our part, we propose another synthetic route for fenfluramine (S)-1 starting from the azide (S)-5 which does not go via the primary amine (S)-6 (Schema 5). 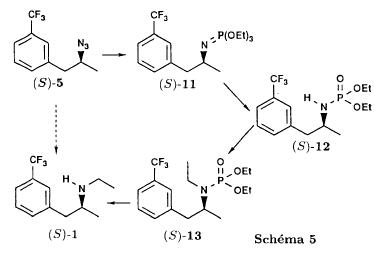 The reaction of Staudinger, reacting a stoichiometric quantity of triethylphosphite on the azide (S)-5 in THF at room temperature [29], gives quantitative yields of the phosphorimide 11 in 48 hours. It's total conversion into the phosphoramide 13, by reacting with ethyl iodide [30] could not be realised [9]. We always obtained different mixtures of the phosphoramides 12 and 13 (referential compounds prepared from the amines 6 and 1). We also noted that the phosphorimide 11 can't be isolated. When the solvent is evaporated, a partly transformation into the phosphoramide 12 takes place. This transformation is completed in less then 2h by simple heating to 100°C under argon after evaporation of the solvent. Because the phosphorimides are strongly basic compounds, we believe that an intramolecular arrangement of the phosphorimide, pictured in Schema 6, takes place. 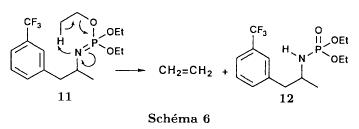 Having the phosphoramide 12, we investigated the alkylation into the phosphoramide 13 in DMF at room temperature [31, 32]: one deprotonates with sodium hydride then alkylates with diethyl sulfate. After treatment with hydrogen bromide [33], one obtains fenfluramine 1 with a yield of 85% and a purity of 97% (GC). With the goal of simplifying the reaction scheme by avoiding the isolation of the intermediates we have again studied the transformation 5 -> 11 -> 12 in DMF (Schema 7). First, we noted that the reaction of Staudinger can be directly realised in this solvent. Thereafter we pinned down the transformation of the phosphorimide 11 into the phosphoramide 12 by reaction with water [34]. One then proceeds as described above. The transformation is thus performed without isolation of a single intermediate with a yield of 83%. 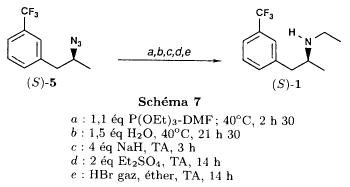 HPLC analysis on silica column of the camphanyl derivate of the amine (S)-1 [22] shows that the optical centre is conserved during the whole transformation. Synthesis via the intermediate 2-chloro-1-[3-(trifluoromethyl)phenyl]propane 14 The yeast reduction of the ketone 2 gives the alcohol (S)-3, of which the authors have inverted the configuration to get the pharmacological active S enantiomer of fenfluramine [7, 8]. Independent research, using the epoxidation method of Sharpless [9, 35] lead us too to the alcohol (S)- 3 which we tried to convert into fenfluramine (S)-1 using a different method. The reaction scheme we kept uses the chloride 14 and proceeds via two inversions of the optical centre (scheme 8). Not owning enough alcohol (S)-3 during the studies, we tested the principle starting with the alcohol (R)-3, produced earlier, and studied the transformation into the azide (R)-5 (scheme 8), the latter being able to lead to (R)-fenfluramine using different methods, like the one outlined above: 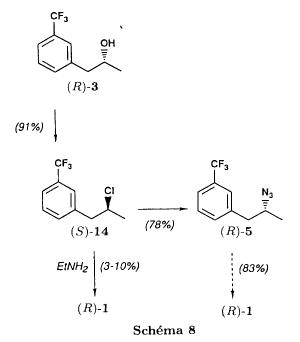 It is well known that the action of thionylchloride on an optically active alcohol gives the corresponding chlorine derivate, with inversion of the configuration in presence of bases and with retention of the configuration in the other case. We have performed the reaction with a catalytical amount of pyridine. One thus obtains the chloride (S)-14 with 91% yield and a purity of 91% (GC): it contains 9% of the elimination products 9, 10Z and 10E which are not separable by chromatography on silica. The direct substitution of the chloride 14 with ethylamine with similar conditions to those used for the mesylate (R)-4b (EtNH2, DMSO, 110°C, 5h30 or EtNH2 (solvent and reactant), 140°C, 5h), gives mainly the elimination products. The yield of fenfluramine is below 10%. By action of sodium azide in DMSO, on the other hand, one obtains the azide (R)-5 with a yield of 78%, the elimination products formed here or in the last step can be removed by chromatography on silica. HPLC analysis on silica of the camphanyl derivate of the amine (R)-6 [22] obtained by catalytic reduction of the azide (R)-5 has confirmed the double inversion without racemisation after comparison with the starting alcohol (R)-3. Then the fenfluramine (R)-1 is prepared without racemisation with a 83% yield starting from the azide (R)-5 like detailed above. This procedure with two inversions allows to transform the alcohol 3 in the azide 5 with the same configuration in two steps with a global yield (non optimised) of 70% and without racemisation. It's thus preferred over the recently published one [7, 8], which needs 5 steps for a lower global yield (55%) and in addition features an epimerisation of 10% [8]. It's a promising way to fenfluramine (S)-1 starting from the alcohol (S)- 3. Conclusion We described a new method allowing the preparation of the (R) or
(S)-1-[3-(trifluoromethyl)phenyl]-propan-2-ols 3 starting from
optical active methyloxiranes and showed the importance of both
enantiomers of this alcohol for the synthesis of (S)-fenfluramine
1. Further we have proposed a method for the preparation of (S)-fenfluramine starting with the alcohol (S)-3 in three steps including a double inversion via the chloride derivate (R)-14 All reactions were realised without racemisation. They could be applied to other analogous structures. Experimental Part General notes The 1H NMR spectra where recorded on a Perkin Elmer R12 (60MHz) or a Bruker AW 80 (80MHz), the 13C spectra on a Varian CFT 20 (20MHz). The chemical shifts are expressed in ppm. Unless otherwise stated, the solvent is deutero-chloroform and the internal reference TMS. Concerning the 13C spectra: the used spectrometer did not allow proton and fluorine decoupled spectra at the same time. The carbon of the CF3 group (q, 1JCF=271.1 Hz) and the carbon on alpha of the CF3 group (q, 2JCF = 32.3 Hz) are barely or not at all visible. On the other hand, the two carbons in beta of the CF3 group are very characteristic (q,3JCF = 3.9 Hz). The IR spectra were recorded on a Perkin Elmer 377. The optical rotation was measured using a Perkin Elmer 241 polarimeter. The microanalyses were performed by the microanalysis service of the INSA in Rouen. The gas chromatographs were performed on a Girdel series 30 chromatograph with a flame ionisation detector. We used a column: 30% SE 30 + 1% triethanolamine on chromosorb W AW 80-100 mesh (0.75m) (maximum usage temperature: 150°C) The liquid chromatographs were realised either with a Chromatem
800 HPLC apparatus of Touzart and Matignon consisting of two
Chromatem 380 pumps and a Shimadzu SPD-2A variable wavelength
detector, or with a Beckman HPLC apparatus consisting of a Beckman
110B pump and a Beckman 160 fixed wavelength detector (254 nm). We
used a Lichrosorb Si60 (5um) Merck (25 cm x 4.6 mm) column with the
following eluants: The composition of the mixtures was measured with an Shimadzu C-R1B integrator. The TLCs were realised on Merck Kieselgel 254GF plates of 0.25mm thickness. They were developed by UV (254nm) or an ethanolic vanilline solution. The silica used for purification is a new Merck Kieselgel 60 230-400 mesh. It was used as is. The anhydrous THF and ether were distilled over sodium and benzophenone right before use. The DCM and the chloroform were distilled and filtered over basic alumina. The DMSO and the DMF were dried over potash, distilled over barium oxide and finally stored over molecular sieves 3A. The acetonitrile was dried and distilled over calcium hydride. None of the presented reactions has been optimised. Reference products - 1-Methyl-2-[3-(trifluoromethyl)phenyl]ethylamine 6 This product was supplied by the Oril society. - N-ethyl-1-methyl-2-[3-(trifluoromethyl)phenyl]-ethylamine 1 This product was supplied by the Oril society. - [1-methyl-2-(3-[trifluoromethyl]phenyl)ethyl]-diethylphosphoramidate 12 To a solution of the amine 6 (2,05g; 10.1 mmol) in DCM (20 ml)
is added triethylamine (1.12g; 1.1 eq.) and three minutes later
diethylphosphorochloridate (1.53ml; 1.05 eq.) (the reaction heats
up). The reaction is stirred for 24h at room temperature. 1N Sodium
hydroxide (25 ml) is added, and the phases separated after shaking.
The aqueous phase is reextracted with DCM (20 ml). The combined
organic phases are washed with 1N HCl (25 ml) and water (10 ml) and
dried over magnesium sulfate. After evaporation, the raw product is
filtered through 5g silica (eluant: 150 ml ether). One such obtains
the phophsoramide 12 (3.0g; yield 88%) - Ethyl-[1-methyl-2-(3-[trifluoromethyl]phenyl)ethyl]-diethylphosphoramide 13 It was prepared in the same way as the phosphoramide 12,
starting with the amine 1 (yield: 91%) - Methyloxirane (R)-7 We used commercial product sold by Fluka. The enantiomeric purity was evaluated to be 82% (+/-5%) using 80Mhz 1H NMR: To 0.5 ml CDCl3 containing 3% TMS is added 23 mg epoxide (R)-7 and 103 mg europium (III) tris(heptafluoropropylhydroxymethylene camphorate) (Eu(hfc)3). The signal of one of the protons of the methylene is thus split in two: 3.15 (d, 3H); 5.60-5.80 (m, 1H); 6.14 (t, 0.09H); 6.34 (t, 0.91H); 6.75-6.90 (m, 1H); i.e. a 82% enantiomeric purity. - 1-[3-(Trifluoromethyl)phenyl]propan-2-ol (R)-3 To fine magnesium turnings (2.16 g, 88.9 mmol) in ether (20 ml) is added a iodine crystal and few ml of a solution of 1-bromo-3- (trifluoromethyl)benzene (20 g; 88.9 mmol) in ether (30 ml). The flask is kept in a hot water bath until the reaction starts. Stirring is started and the rest of the solution is added during 2h30 maintaining the temperature between 34-36°C. After the addition, the vial is rinsed with some ether (5 ml) and the stirring maintained for further 30 min at room temperature before dosing [36] (titer = 1.37N) To cupric bromide (0.34 g; 2.4 mmol; 0.15 eq; 99.999% Aldrich
Gold Label) in ether (20 ml) at -30°C is added during 20 min
the solution of the magnesium compound 8 in ether (25.7 ml; 35.3
mm, 1.5 eq.) prepared above and the reaction stirred for 5 min at
-30°C. One then adds during 5 min using a syringe the
methyloxirane (R)-7 (1.65 ml; 1.0 eq; 82% ep). One then lets
the temperature rise to 0°C and maintains for 3 hours at
0°C, then pours on a mixture of ice (50 g) and hydrochloric
acid 3N (50 ml). The phases are separated and the aqueous phase is
reextracted with ether (2 x 100 ml). The combined organic phases
are dried over magnesium sulfate, filtered and the solvent
evaporated. The product is distilled (bp = 102-103°C/13-14 mm
Hg), then chromatographed over silica (eluant: PE-ether 100:9 to
0:100) giving the alcohol (R)-3 (4.34 g; 90% yield; 93%
GC) 2 step synthesis of fenfluramine - 2-[Methylsulfonyloxy)-1-[3-(trifluoromethyl)-phenylpropane (R)-4b To the alcohol (R)-3 (1.19g; 5.8 mmol; 80% ep) in
pyridine (6 ml) at 0°C is added drop by drop in 2 minutes mesyl
chloride (0.74g; 1.1 eq). The reaction is stirred for 22h30 at room
temperature, the pyridine evaporated and the residue taken up in
DCM (30 ml). The solution is washed with 1N hydrochloric acid (15
ml), a saturated sodium hydrogen carbonate solution (15ml) and
water (15ml), dried over magnesium sulfate, filtered and the
solvent evaporated. The product (1.48 g) is chromatographed over
18g silica (eluant: PE-ether 100:0 to 75:25) giving the mesylate
(R)-4b (1.37 g; 83% yield) containing about 5% alcohol
(R)-3 (1H NMR). - 2-(Trifluoromethylsulfonyloxy)-1-[3-(trifluoro-methy)phenyl]propane (R)-4c To trifluoromethanesulfonic anhydride (0.35 ml; 1.05 eq) in DCM
(2 ml) at 0°C is added drop by drop the alcohol (R)-3
(0.41 g; 2.0 mmol; 80% ep) and pyridine (175 mg; 1.1 eq) in DCM (1
ml) over the course of 5 min. The reaction is stirred for 2h at
0°C. Water (5 ml) is rapidly added, then DCM (10 ml). The
phases are separated and the organic phase washed with 1N
hydrochloric acid (10 ml), saturated sodium hydrogencarbonate
solution (10 ml) and water (10 ml), dried over magnesium sulfate,
filtered and the solvent evaporated. One gets the triflate
(R)-4c (0.63 g; 92% yield). This product is unstable and
thus used right away. - Substitution of the mesylate by ethylamine: typical procedure in DMSO To the mesylate (R)-4b (364 mg; 1.29 mmol; prepared
starting from the alcohol (R)-3 with 80% ep) in DMSO (1 ml)
at 22°C in an autoclave is added anhydrous ethylamine (0.4 ml;
about 4 eq.). One heats to 100-105°C during 3 h, then lets
cool. 3N Hydrochloric acid (3 ml) is added and the reaction
extracted with ether (2 x 10 ml). The combined organic phases are
washed with water (5 ml), dried over magnesium sulfate, filtered
and the solvent evaporated. One such obtains the fraction of the
"non animated byproducts" which is analysed by GC (identification
by comparison to the reference products in our possession). One
extracts in the same manner the aqueous phase basified with 10N
sodium hydroxide (1 ml). One thus obtains the amine (S)-1
(121 mg; 41% yield; 90% pure on GC). - Typical procedure in ethylamine In an autoclave, the mesylate (R)-4b (386 mg; 1.37 ml; prepared from the alcohol (R)-3 with 80% ep) in anhydrous ethylamine (2 ml) is heated to to 90°C for 16 h. One cools, opens the autoclave and lets the ethylamine evaporate under the fume hood. The residue is taken up in 5N sodium hydroxide (5 ml), shaken and extracted with ether (3 x 10 ml). The combined organic phases are extracted with 3N hydrochloric acid (2 x 3 ml), filtered and evaporated. One such obtains the byproducts. The combined aqueous hydrochloric acid phases are made basic with 10N sodium hydroxide (2.5 ml) and extracted with ether (3 x 10 ml). The combined organic phases are washed with water (5 ml) and dried over magnesium sulfate. After evaporation, one obtains the amine (S)-1 (152 mg; 48% yield; >99% GC) [alpha]24/D = +7.8 (c = 7.90; absolute ethanol) (ep: 89%). - Substitution of the triflate by ethylamine To the triflate (R)-4c (0.63 g; 1.87 mmol; prepared from the alcohol (R)-3 with 80% ep) in acetonitrile (8 ml) at -20°C is added during 3 min anhydrous ethylamine (0.5 ml, about 4 eq.) in acetonitrile (2 ml) preferably cooled. One maintains for 4 h at -20°C than lets the temperature climb to room temperature. The solvent is evaporated, the residue taken up in 5N sodium hydroxide (6ml) and vigorously shaken. One extracts with ether (2 x 20 ml). The combined organic phases are extracted with 3N hydrochloric acid (5 ml), washed with water (10 ml), dried over magnesium sulfate, filtered and evaporated. These byproducts are analysed with GC. The combined aqueous hydrochloric acid phases are made basic with 10N sodium hydroxide (4 ml) and extracted with ether (2 x 20ml). The combined organic phases are washed with water (10 ml), dried over magnesium sulfate, filtered and evaporated. One thus obtains the amine (S)-1 (0.26 g; 60% yield; > 99% GC) [alpha]24/D = +7.5 (c = 7.86; absolute ethanol) (ep: 85%). Remark: While doing experiments with racemic compounds, we
omitted the treatment with 5N sodium hydroxide and obtained a white
solid which has been characterised as being the
trifluoromethanesulfonate of the amine (yield: 15 to 40%, depending
on the experiment) Synthesis of the azide 5 - 2-chloro-1-[3-(trifluoromethyl)phenyl]propane (S)-14 To the alcohol (R)-3 (684 mg; 3.1 mmol; 80% ep) in
chloroform (1.5 ml) is added thionyl chloride (0.4g; 1.1 eq) and a
drop pyridine. The mixture is refluxed with heavy stirring for 7 h
30. The low boiling products are evaporated and the residue taken
up in DCM (10 ml). One washes with water (5 ml), saturated sodium
bicarbonate solution (5 ml) and water (5 ml), dries over magnesium
sulfate and evaporates the solvent. The thus obtained product is
filtered through 5g silica (eluant: petroleum ether) to give the
chloride (S)-14 (682 mg; raw yield = 91%; 91% GC (9%
elimination products)). - 2-Azido-1-[3-(trifluoromethyl)phenyl]propane 5 : Azide (R)-5 from the chloride (S)-14 To the chlorine (S)-14 (0.66 g; 3.0 mmol; prepared from
the alcohol (R)-3 with 80% ep) in DMSO (4 ml) is added
sodium azide (0.38 g; 2eq). One heats to 75°C for 8 h. One lets
return to room temperature, adds 0.1N hydrochloric acid (10 ml) and
extracts with ether (3 x 10 ml). The combined organic phases are
washed with water (5 ml) and dried over magnesium sulfate. After
evaporation of the solvent, the raw product (3.73 g) is
chromatographed on 8 g silica (eluant: pentane) to give the azide
(R)-5 (0.53 g; 78% yield; 95% GC (contains 3% of the
chloride (S)- 14)) In order to determine the enantiomeric purity, a part of the
product has been reduced to the amine in the following way: to the
azide (R)-5 (258 mg; 1.13 mmol) in THF (5 ml), one adds 5%
palladium on calcium carbonate (26 mg; 10% per weight). Under heavy
stirring, a temperature of 21°C and a standard pressure of
hydrogen are maintained for 18h. The catalyst is filtered off and
rinsed with THF (10 ml) and the solvent evaporated. The raw product
(189 mg) is distilled (bp = 90-95°C/15 mm Hg) to give the
colourless amine (R)-6 (155 mg; 67% yield; 95% GC). - Azide (S)-5 from the mesylate (R)-4b To the mesylate (R)-4b (0.39g; 17 mmol; prepared from the
alcohol (R)-3 with 80% ep) in DMSO (2ml) is added sodium
azide (135 mg; 1.5 eq). The mixture is heated for 30 min at
70°C, then cooled. One adds 0.1N hydrochloric acid (5 ml) and
extracts with ether (2 x 10 ml). The combined organic phases are
washed with water (5 ml), dried over magnesium sulfate, filtered
and the solvent evaporated. The raw product (309 mg) is filtered
through 5 g silica (eluant: petroleum ether) to give the azide
(S)-5 (248 mg; 78% yield; 99% GC) lambda (nm): 589 578 546 436 365 HPLC analysis of the camphanylated derivate of the amine (S)-6 (obtained by catalytic reduction): S/R = 90/10 (ep: 80%) Remark: Trials with bigger amounts (> 5g) of the racemic products gave yields greater or equal to 95% Synthesis of fenfluramine 1 starting from the azide 5 To the azide 5 (207 mg; 0.903 mmol) in THF-d8 (1 ml), is added
ethyl phosphite (0.19 ml; 1.0 eq). After 40 h at 26-29°C the
spectra are recorded (after adding a drop of TMS): When the reaction is done in the same manner in ether or THF, but the solvent evaporated, the 1H NMR and the 13C NMR (in CDCl3) show that a mixture of the products 11 and 12 of indeterminable composition is obtained. - Pinning down the ethylation of the phosphoramide 12: typical procedure To the racemic phosphoramide 12 (331 mg; 0.976 mmol) in DMF (2.5 ml) at 24°C is added at once sodium hydride (86%; 94 mg; about 3.5 eq). After stirring for 1 h 30, diethyl sulfate (0.26 ml; 2 eq) is added at once (exothermic reaction). The reaction is stirred at 24°C over night. One slowly adds water (5 ml) and extracts the aqueous phase with ether (2 x 20 ml). The combined organic phases are washed with water (10 ml) dried over magnesium sulfate, filtered, concentrated to 5 ml and slowly gassed with hydrobromic acid for 10 min. After standing over night at room temperature, the solvent is evaporated and the residue taken up in 5N sodium hydroxide (5 ml). One extracts with ether (2 x 15 ml). The organic phases are washed with water, dried over magnesium sulfate, filtered and the solvent evaporated. One obtains the amine 1 (192 mg; 85 % yield; 97% GC (amine 6 < 1%)). - Transformation of the azide 5 into the amine 1 through the intermediate of the phosphoramides 12 and 13 * Case of the azide (R)-5 (prepared from the * alcohol (R)-3 at 80% ep, via the chloride * (S)-14) To the azide (R)-5 (254 mg; 1.11 mmol) in DMF (1 ml) at
room temperature is added ethyl phosphite (0.25 ml; 1.1 eq) and the
mixture heated to 40°C. After 2 h 30, water (30 ul; 1.5 eq) is
added and the stirring continued at 40°C during 21 h 30. One
weights the sodium hydride (86%; 134 mg; 4eq), rapidly adds about a
half and after 15 minutes the second half. One maintains 2 h 30 at
room temperature. One then adds diethyl sulfate (0.29 ml, 2 eq)
(exothermic reaction) and stirs at room temperature over night.
Carefully water (5 ml) is added and the workup done like in the
previous example. One obtains the amine (R)-1 (213 mg; 83$
yield; 97% GC (amine (R)-6: 1.5%)). * Case of the azide (S)-5 (prepared from the * alcohol (R)-3 at 92% ep, via the mesylate * (R)-4b) Using the same procedure, starting with 273 mg of the azide
(S)-5 we obtained 229 mg of the amine (S)-1 (yield =
83%). After distillation (yield = 47%), on obtains: GC purity = 86%
(amine (S)-6: 11%). Références
Bull. Soc. Chim. Fr. (1993) 130, 459-466 BP 118 76134 Mont-Saint-Aignan Cedex, France Adresse actuelle: Institut de Recherches Servier, 11, rue des Moulineaux, 92150 Suresnes, France Société ORIL, 13 rue Auguste Desgenétass, 76210 Bolbec, France (reçu le 30 octobre 1992, accepté le 28 mars 1993) Summary - Epoxides, amino alcohols, and aziridines as key intermediates in the asymmetric synthesis of (S)-fenfluramine. The epoxides 3E, 3Z and 8 were obtained from the isomeric alkenes 2E, 2Z and 7. The epoxides were ring-opened by ethylamine in ethanol yielding mixtures of the amino alcohols 4E and 11E, 4T and 11T, and 9, respectively, which were transformed into the aziridines 5trans, 5cis and 8 were reduced regiospecifically into 1-[3-(trifluoromethyl)phenyl]propan-2-ol 6, a potent precursor to fenfluramine 1. The validity of our method for asymmetric synthesis has been demonstrated by a synthesis of (S)-fenfluramine 1 starting from the amino alcohol (S)-9. fenfluramine / asymmetric synthesis / epoxide / amino alcohol / aziridine / regiospecific catalytic hydrogenation / 1-[3- (trifluoromethyl)phenyl]propan-2-ol Introduction During our studies on the asymmetric synthesis of (S)-fenfluramine 1 [1], we investigated the following scheme, using classical reactions and simple intermediates: alkenes, epoxides, aminoalcohols and aziridines: 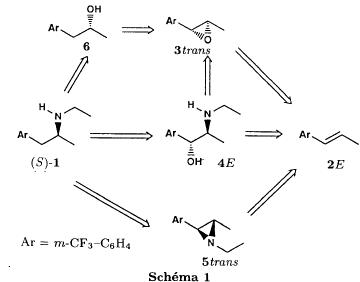 The asymmetric center can for example be introduced using the alkene 2E using enantioselective epoxidation. This method is subject of lots of research and the yields are constantly improved [2]. The such obtained epoxide 3trans is then subjected to the action of a carefully selected nucleophile in order to give the enantiomerical pure aminoalcohol 4E. Hydrogenolysis of the hydroxyl in benzylic position will give (S)-fenfluramine 1. The aminoalcohol 4E and the aziridine 5trans could also be obtained starting with the alkene 2E using enantioselective hydroxyamination [3, 4] or enantioselective aziridination. This latter method, studied by multiple teams in the last years [5, 6] deserves being considered, because encouraging results have been obtained recently [7]. Another alternative would be the regiospecific reduction of the epoxide 3trans to the alcohol (R)-6, of which we have shown before that it can be used as precursor for (S)- fenfluramine in multiple processes. We studied some of these possibilities starting with the alkenes 2E, 2Z and 7. Before embarking upon the asymmetric epoxidation, hydroxyamination and aziridination of these alkenes, we realised the synthesis of all compounds of the racemic series (Schema 2). The results of these preliminary studies, which allowed us to characterise the different intermediates and to study the limits of each reaction, are presented in this paper. Then we studied the synthesis of (S)-fenfluramine 1 via the intermediate aminoalcohol (S)-9. Results and discussion Starting with the isomeric alkenes 2E, 2Z and 7 we have investigated three parallel reaction paths (Schema 2). We have pictured the optical active products, only which are of interest for the asymmetric synthesis of (S)-fenfluramine, but all the presented reactions were realised using racemic products, with the exception of the transformation 9 -> 12 -> 1. 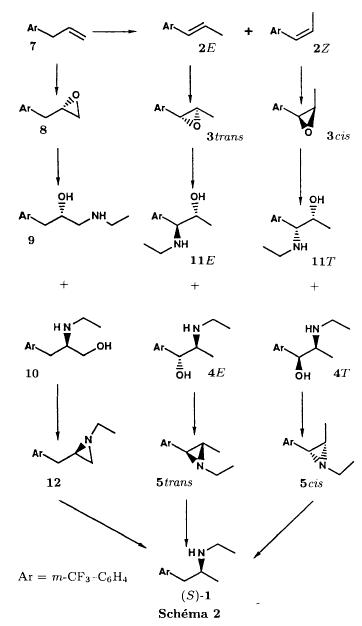 Preparation of the epoxides and the aminoalcohols The alkene 7 is obtained by condensation of 3-(trifluoromethyl)phenyl magnesium bromide with allyl chloride in refluxing ether, then isomerised by the action of sodium hydroxide in refluxing n-butanol into a mixture of the alkenes 2Z and 2E (2E/2Z : about 85/15), which are separated by distillation. The epoxides 3trans, 3cis and 8 were obtained by reacting 3-chloroperoxybenzoic acid (mCPBA) with the corresponding alkene and purification by distillation. According to the literature [9] it seems rather difficult to obtain uniform ring opening of the epoxides favouring the aminoalcohols 4 and 10. In a first attempt we reacted the epoxides with ethylamine in ethanol during two days at 70°C, hoping to obtain and characterise the different aminoalcohols 4, 9, 10 and 11. Starting with the epoxide 8 we have isolate the alcohol 9, pure according to GC, with a yield of 97% without purification. The regioisomer aminoalcohol 10 could not be detected. The epoxide 3trans gives the aminoalcohols erythro 4E and 11E (4E/11E: 24/76) with 82% yield. The epoxide 3cis gives the epoxides threo 4T and 11T (4T/11T: 93/7) with 86% yield. This result was obtained by extrapolation because the sample used contained 8% of the epoxide 3trans. The products were separated by chromatography on silica. The configurations could be revealed using the 1H NMR coupling constants between the protons attached to the carbons attached to the heteroatoms and by comparison to the aminoalcohols with related structure [10]. 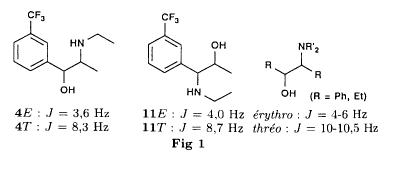 The results show an inverse regioselectivity for the epoxides 3trans (4E/11E: 24/76) and 3cis (4T/11T: 93/7). Using the epoxide 3cis, one obtains better regioselectivity and further it gives the preferred product 4T. Ring opening in alpha position to the aromatic group predominates only in the case of the isomer 3trans where the conjugation of the aromatic group with the neighbouring carbon, carrying the oxygen, is possible. 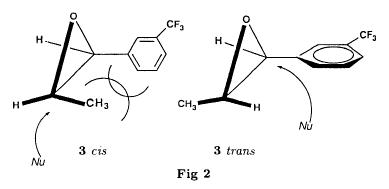 Try at the hydrogenolysis of the benzylic hydroxyl function of the aminoalcohols 4 According to the literature [11], hydrogenation of an alcohol group in benzylic position is easy and is performed in mild conditions (5% Pd/C in alcohol at room temperature and standard pressure). However beta-aminated benzylic alcohols are much harder to hydrogenolyse: the yield is acceptable when the reaction is performed in acidic medium [12-14]. Our experiments, using a mixture of the aminoalcohols 4E + 4T + 11E + 11T and different conditions were not successful: when using mild conditions (ethanol-60°C-24h without activator), the reactants were isolated unchanged, and using harder conditions (acetic acid or water-100-110°C-1 to 16 h-in presence of concentrated hydrochloric and sulfuric acid), gives general degradation. The expected products, the amine 1 and the alcohol 6 could only be detected in trace amounts. We believe that the trifluoromethyl group renders the cutting in benzylic position difficult by deactivating the aromatic ring. The regioselective opening of the epoxides on one hand and the difficulties of the hydrogenolysis of the aminoalcohols 4 and 11 on the other hand made us study a new strategy, going via the aziridines 5 and 12. One notes that the 2 aminoalcohols 4E and 11E give the same aziridine 5trans, likewise the aminoalcohols 4Z and 11Z giving the aziridine 5cis. A given epoxide is transformed into the aziridine with opposite configuration (see scheme 2). Preparation of the aziridines One finds in the literature multiple methods giving aziridines starting from aminoalcohols [15-19]. They all use phosphorus compounds, with the exception of the method by Wenker [19] which uses concentrated sulfuric acid in very hard conditions. Using phosphorus compounds, the transformation is realised in one step in mild conditions. One can use dibromo-triphenylphosphine in presence of triethylamine [15], triphenylphosphine in presence of triethylamine and carbon tetrachloride [16], tretraiodophoshporus [17] and (diethoxy triphenyl)phosphorane [18]. In two cases [15, 18], the conservation of the chiral centres by Walden inversion has clearly been established. The method we retained used the simplest and most easily accessed reagents: triphenylphosphine, triethylamine and carbon tetrachloride [16]. Starting with the aminoalcohols 4, 11 and 9 we thus obtained the aziridines 5cis, 5trans and 12, which have never been described before. The yields after purification by distillation are between 70 and 75%. For the preparation of the aziridine 12 we have also carried out multiple experiments with triethyl phosphite. The yield is a bit less (60%). This variation of the original method [16] has never been described as far as we know. Going via these aziridines has the following advantages: it is unnecessary to separate the aminoalcohols 4 and 11, and the useless aminoalcohol 11 is such recycled in the reaction without any additional work. It is also a quick method for the valorisation of the aminoalcohol 9. Further, it is no longer necessary to bother with the regioselectivity of the nucleophilic attack on the epoxide, which allows the usage of the simplest method for the preparation of the aminoalcohols: ethylamine in ethanol. Reduction of the aziridines It remained to work out the transformation of the aziridines 5 and 12 into fenfluramine 1. The aziridines 5 can lead to the amines 1 and 13, the aziridine 12 to the amines 1 and 14 [20] (scheme 3). 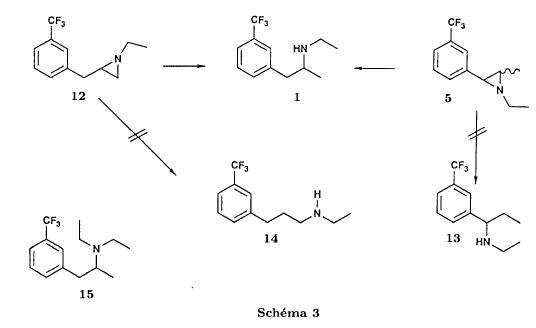 In order to realise the ring opening of the aziridines 5 and 12 into fenfluramine 1, we used catalytic hydrogenation [11], peculiar to open the ring at the less substituted side, in the case of the aziridine 12, and benzylic hydrogenolysis in the case of the aziridines 5. The experiments were performed in ethanol at standard temperature and pressure (table I).
We noted that in all three cases the reaction is perfectly regioselective, the amines 13 and 14 could not be detected. The only observed side reaction is the formation of the diethylamine 15, especially when using palladium on calcium carbonate. We believe that, on contact with the catalyst, the ethanol is dehydrogenated into acetaldehyde [21] which gives the diethylamine by amination of the amine 1 formed during the reduction. Provided that the regioselectivity is preserved, the replacement of ethanol by another solvent should allow to obtain the pure amine 1. Usage of palladium on carbon minimises the formation of diethylamine 15 in the case of the aziridines 5trans and 12. On the other hand, the aziridine 5cis is less reactive and needs a reaction time of 3 days instead of one. Therefore the byproduct is present in non deniable amounts. Reduction of the epoxides The epoxide 3 can lead to the alcohols 6 and 16 and the epoxide to the alcohols 6 and 17. 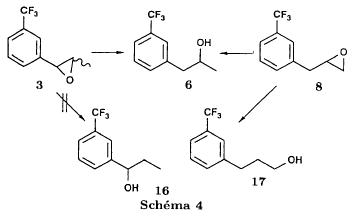 Only the alcohol 6 is a potential precursor of the fenfluramine 1. Given the satisfying results obtained using the aziridines, we realised the same type of study (table II). The alcohol 16 was never detected. In the case of the epoxide 8, the alcohol 17, formed when using palladium on carbon, disappears when replacing the latter by palladium on calcium carbonate. Among the byproducts one finds the deoxygenated product 18, when using palladium on carbon and the ketone 19, formed by dehydrogenation of the alcohol 6 on contact with the catalyst [21], particularly when using palladium on calcium carbonate. 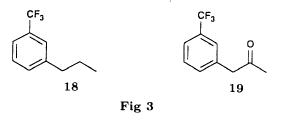 Thus, like in the case of the aziridines, we were able to obtain a regioselective ring opening of the epoxides 3 and 8 by hydrogenolysis of the bond in benzylic position using palladium on carbon in the case of the epoxide 3 and by hydrogenation of the ring on the less substituted side by palladium on calcium carbonate in the case of the epoxide 8. Asymmetric synthesis of the (S)-fenfluramine From a stereochemical viewpoint, one could contemplate to access the many proposed precursors of the amine 1 (epoxide, aminoalcohol or aziridine) by totally different reactions than those we have described. Thus the validity of our reaction scheme for the asymmetric synthesis of (S)-fenfluramine has been shown starting with the aminoalcohol (S)-9. The latter was prepared using the epoxyalcohol (1R,2R)-20, which in turn was obtained by asymmetric epoxidation of the corresponding allyl alcohol according to the method of Sharpless and transformed into fenfluramine via the intermediate aziridine (R)-12 (schema) using the technique described above. 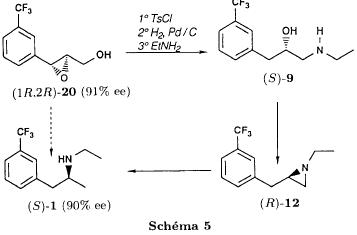 The comparison of the enantiomeric excesses of the epoxyalcohol 20 and the fenfluramine (S)-1 shows that there is no racemisation during the different reactions, notably during the cyclisation into aziridine. The detailed results of these transformations will be published later [22]. Conclusion Regarding synthesis, we have prepared and characterised a number of intermediates: racemic epoxides, aminoalcohols and aziridines, and shown possible synthetic pathways to fenfluramine 1 using simple methods starting with the alkenes 2Z, 2E and 7. Regarding stereochemistry, we have shown the validity of our reaction scheme to prepare (S)-fenfluramine starting with the aminoalcohol (S)-9. The enantioselective epoxidation, hydroxyamination and aziridination reactions can now be approached. From a general point of view, given the large applicability of the presented reactions, the regiospecificity of the hydrogenation reactions and the hydrogenolysis, we believe that this work gives new perspectives for the synthesis of 2-(alkylamino)-1-arylpropanes and 1-arylpropane-2-ols which are potential precursors. Experimental part General indications The 1H NMR spectra were recorded on an Perkin Elmer R 12 (60 Mhz) or a Bruker AW 80 (80 MHz), the 13C NMR spectra on a Varian CFT 20 (20 MHz). The chemical shifts are expressed in ppm. Unless otherwise noted, the solvent is deuterochloroform and the internal reference is TMS. Concerning the 13C NMR spectra: the used spectrometer did not allow the recording of fluorine and proton decoupled spectra at the same time. The carbon of the CF3 group (q, 2JCF=271.7 Hz) and the carbon on alpha to the CF3 group (q, 2JCF=32.3 Hz) are only hardly or not at all visible. They were never indicated. On the other hand, the 2 carbons beta to the CF3 group are very characteristic (q, 3JCF=3.9 Hz). The IR spectra were recorded using a Perkin Elmer 377. The melting points were measured using a melting point microscope Reichert Jung. The microanalyses were performed by the common microanalysis service of the INSA of Rouen. The gas chromatographs were realised with a Girdel series 30
chromatograph with a flame ionisation detector. The used columns
are: The percentages of the components were measured using a Shimadzu C-R1B integrator. The TLCs were realised on silica plates Merk Kieselgel 254GF with 0.25mm thickness. They were developed using UV (254 nm) or an ethanolic vanilline solution. The silica used for purification is a Merck Kieselgel 60 230-400 mesh. It was used as is. The anhydrous ether was distilled over sodium and benzophenone right before use. The dichloromethane was distilled and filtered over basic aluminium. The acetonitrile was dried and distilled over calcium hydride. The RPE anhydrous ethanol (Carlo Erba) was used as is. None of the described reactions was optimised. Preparation of the alkenes - 3-[3-(Trifluoromethyl)phenyl]propene 7 The magnesium compound is prepared liked previously described
[8] using 3-(trifluoromethyl)bromobenzene (50.2 g; 223 mmol) and
fine magnesium turnings (5.55 g; 228 mmol). After 30 min at room
temperature, the reaction is newly refluxed. Allyl chloride (17.6
g; 230 mmol) in ether (30 ml) is then added over 45 min. The
reaction is refluxed for 2h then cooled and poured on ice (65g).
The flask is rinsed with water (20 ml). The phases are separated
and the aqueous phase extracted with ether (45 ml). The combined
organic phases are dried over magnesium sulfate, filtered and
evaporated. After distillation (bp = 54-54°C/13-14 mm Hg), the
alkene 7 (33.8 g; 81% yield) is obtained. - 1-[3-(trifluoromethyl)phenyl]propene 2 cis and 2 trans Sodium hydroxide (392 mg 9.8 mmol) is added to 3-[3-(trifluoromethyl)phenyl]propene (33.1 g; 178 mmol) in n-butanol (33 ml). The reaction is refluxed for 7h, cooled and tranfered into a separatory funnel. It is washed with 3N hydrochloric acid (4 ml), water (2 x 3.5 ml), and distilled: first the n-butanol then the products (bp = 66-70°C/13-14 mm Hg). A mixture of both isomers is obtained (31.1g; 94% yield; cis/trans = 13/87 according to GC). In order to separate both isomers, we have distilled 21g of the mixture at 15 mmHg through a Nester Faust spinning band column and obtained 3 main fractions: 63°C <= bp <= 65°C: 1.85g (yield = 9%) GC: 89%
2cis Alkene 2cis (1st fraction): bp = 63-65°C/15
mm Hg. GC purity = 89% (8% 2trans and 3%
impurities) (column B) Alkene 2trans (3rd fraction): bp = 73°C/15
mm Hg. GC purity = 98.4% (1.6% 2cis and impurities
< 0.1%) (column B) Preparation of the epoxides - 2-[3-(Trifluoromethyl)benzyl]oxirane 8 Meta-chloroperoxybenzoic acid (80%; 5.8g; 27.0 mmol) is added
over 5 min to dichloromethane (25 ml) at 20°C in small
aliquots. The reaction is stirred for 5h30 at 20°C.
Dichloromethane (20 ml) is added and the reaction is washed
successively with 10% aqueous sulfite solution (10 ml), saturated
sodium bicarbonate solution and water (10 ml). The organic phase is
dried over magnesium sulfate, filtered and evaporated. The residue
is taken up in petroleum ether (25 ml) and left standing at room
temperature over night. Then the precipitate is removed by
filtration, the solvent evaporated and the residue distilled (bp =
65-95°C / 13-14 mmHg). The epoxide 8 (2.12 g; 78% yield)
is thus obtained. - 2-methyl-3-[3-(trifluoromethyl)phenyl]oxirane 3 (cis and trans) The epoxides 3cis and 3trans were prepared like previously described, starting with the alkenes 2cis and 2trans. Epoxide 3cis (85% yield; 89% GC (9%
3trans and 2% impurities) (column B)). Bp =
84-86°C/13 mm Hg. Epoxide 3trans (71% yield; 98% GC). bp =
70-90°C/13-14 mm Hg. Preparation of the amino alcohols - 3-Ethylamine-1-[3-(trifluoromethyl)phenyl]propan-2-ol 9 Anhydrous ethylamine (5 ml; 76.6 mmol) is added at once to the
epoxide 8 (2.11 g; 10.5 mmol) in absolute ethanol (50 ml). The
reaction is heated to 70°C during 47h. After evaporation of the
solvent and the excess ethylamine, one obtains the aminoalcohol
9 (2.52g; yield 97%; GC >99%). mp = 43-45°C
(microscope). - 1-Ethylamino-1-[3-(trifluoromethyl)phenyl)propan-2-ol 11 and 2-ethylamino-1-[3-(trifluoromethyl)phenyl]propan-1-ol 4 * With erythro configuration 4E and
11E Aminoalcohol 4E (eluant: ether). mp =
65-67°C (microscope). Aminoalcohol 11E (eluant: ether then
acetone): very thick oil. * With threo configuration 4T and
11T 11: Yield = 11% (about 50% erythro and 50% threo), Aminoalcohol 4T: mp = 54-56°C
(microscope) Aminoalcohol 11T: very thick oil. Preparation of the aziridines - 1-Ethyl-2-[3-(trifluoromethyl)benzyl]aziridine 12 * Method using triphenylphosphine Triethylamine (491 mg; 1 eq), tetrachloromethane (747 mg; 1 eq) and triphenylphosphine (1.46 g; 1.15 eq) are added to the aminoalcohol 9 (1.2 g; 4.85 mmol) in acetonitrile (5 ml). The reaction is heated to 42°C for 3h30. The precipitate is filtered off and washed with acetonitrile (5 ml). The solvent is evaporated and the residue taken up 4 times in petroleum ether (10 ml). The organic phases are combined and the solvent evaporated. The residue is distilled (bp = 70-75°C/0.5 mm Hg) giving the aziridine 12 (817 mg; 73% yield; GC >99%) 1H NMR: 1.00 (t, J=6.7 Hz, 3H); 1.20-1.80 (m, 3H);
1.95-2.55 (m, 2H); 2.75 (d, J=5.3 Hz, 2H);
7.40-7.70 (m, 4H). * Method using triethyl phosphite Triethylamine (208 mg; 1 eq), tetrachloromethane (316 mg; 1 eq) and triethylphosphite (0.49 ml; 1.15 eq) are added to the aminoalcohol 9 (508 mg; 2.05 mmol) in acetonitrile (2 ml). The reaction is heated to 45°C for 5h20. After cooling, first 1N hydrochloric acid (10 ml) then ether (20 ml) are added. The phases are separated and the aqueous phase is washed with ether (2 x 20 ml). It is basified with 10N sodium hydroxide (2 ml) and reextracted with ether (2 x 20ml). The two etherical phases are combined, washed with water, dried over magnesium sulfate, filtered and evaporated. The aziridine 12 (341 mg; 73% yield; GC about 80%) is thus obtained. - 1-Ethyl-2-methyl-3-[3-(trifluoromethyl)phenyl]aziridine 5 We proceeded in the same way as for the aziridine with the triphenylphosphine route. Aziridine 5trans: Reacting the
aminoalcohol 11E (1.27 g; 5.12 mmol) for 6h at
40°C gives after distillation (bp = 105-107°C/14-15 mm Hg)
the aziridine 5trans (882 mg, 75% yield; GC
98%). Aziridine 5cis: Reacting the aminoalcohol
4T (489 mg; 1.98 mmol) for 3h at 45°C gives
after distillation (bp = 93-96°C/15 mm Hg) the aziridine
5cis (317 mg; 70% yield; GC 95%). Reduction of the aziridines: typical procedure Palladium on carbon 5% (35 mg; 10% per weight) is added to the aziridine 12 (332 mg; 1.45 mmol) in ethanol (5 ml). The reaction is stirred for 16 h at 19°C under hydrogen atmosphere at standard pressure. The catalyst is filtered off, rinsed with ethanol (20 ml) and the solvent evaporated. The amine 1 is thus obtained (305 mg; 91% yield; 95% GC). Catalytic reduction of the epoxides - Reference alcohols 16 and 17 * 3-[3-(trifluoromethyl)phenyl]propan-1-ol 17 Palladium on carbon 5% (46 mg; 10% per weight) is added to
3-[3-(trifluoromethyl)phenyl]prop-2-en-1-ol (452 mg) [22] in
ethanol (10 ml). The reaction is vigorously stirred for 26 h at
20°C under hydrogen atmosphere at standard pressure. The
catalyst is filtered off, rinsed with ethanol (20 ml) and the
solvent evaporated. The raw product (342 mg) is chromatographied
over 6 g silica (eluant: petroleum ether-ether 100:0 to 75:25) to
give the alcohol 17 (263 mg; 57% yield; 98% GC). * 1-[3-(Trifluoromethyl)phenyl]propan-1-ol 16 Platinum oxide (4 mg; 1% per weight) is added to
1-[3-(trifluoromethyl)phenyl]prop-2-en-1-ol (407 mg; 2.01 mmol)
[22] in ethanol (5 ml). The reaction is vigorously stirred for 24 h
at 20°C under hydrogen atmosphere at standard pressure. The
catalyst is filtered off, rinsed with ethanol (5 ml) and the
solvent evaporated. The raw product (390 mg) is chromatographied
over 6 g silica (eluant: petroleum ether-ether 100:0 to 95:5) to
give the alcohol 16 (333 mg; 81% yield; 95% GC). - Catalytical reduction of the epoxides The reaction time and the quantity of catalyst were not
optimised.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||