Abstract
α,β-Unsaturated nitroalkenes are readily reduced to the corresponding aldoximes and ketoximes in good yields, using a system of decaborane (B10H14) and DMSO in methanol in the presence of 10% Pd/C at room temperature under nitrogen.
Introduction
Nitroalkenes are valuable precursors to a wide variety of building blocks and intermediates in organic synthesis1. The reduction of nitroalkenes to oximes is important because oximes are latent protected forms of aldehydes and ketones. Representative reports describe the reduction of conjugated nitroalkenes to oximes using a HCOONH4–Pd/C system2, NaH2PO2–Pd/C3, Cr(II)Cl2–HCl4 and Sn–HCl5. These have drawbacks such as low yields and limitation of the resulting ketoxime.2 The low yields are due to side reactions such as ketone formation, dehalogenation and polymerization. Two groups reported efficient reduction conditions using SnCl2·H2O6 and Pb/AcOH–DMF.7 The tin(II)chloride reductions solved the problem associated with dehalogenation. However, selective reduction in the presence of a nitro group8 was not found in the literature. Here, we report an efficient reduction of nitroalkenes to oximes using decaborane as a transfer hydrogenation reagent in the presence of 10% Pd/C as a catalyst and DMSO as an additive.
Results and discussion
Scheme 1.
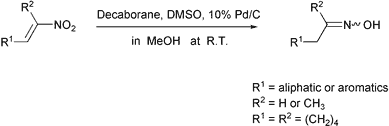
In connection with our recent studies on decaborane as a hydrogen donor in catalytic transfer reactions,9 nitroalkenes in MeOH were found to be smoothly converted into oximes using a system of decaborane and DMSO (dimethyl sulfoxide) in the presence of Pd/C under nitrogen at room temperature as shown in Scheme 1.
Scheme 2.
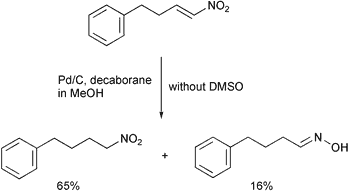
Under the reaction conditions, the reduction of nitroalkenes using 30 weight% of 10% Pd/C, 30 mol% of decaborane and 5 eq. of dimethyl sulfoxide gave the corresponding aldoximes and ketoximes in good to high yields. The results are summarized in Table 1. The addition of DMSO to the reaction mixture dramatically changed the results. To test the efficiency of the reaction in the presence of DMSO, two examples were tried in the absence or presence of DMSO. While the reaction of 5-(2-nitrovinyl)-1,2-methylenedioxybenzene (entry 8) in the presence of 5 eq. of DMSO gave the corresponding oxime in 77% isolated yield, in the absence of DMSO it gave the corresponding oxime in 38% isolated yield. As another example, the reaction of (4-nitrobut-3-enyl)benzene in the absence of DMSO gave an oxime in 16% isolated yield and a nitroalkane in 65% isolated yield (Scheme 2), which has been observed in reductions of nitroalkenes using borohydride reagents,10 However, the reaction in the presence of 5 eq. of DMSO gave an oxime in 71% yield after column chromatography. The poison for the reduction of the nitroalkene into an alkylamine may be a small amount of dimethyl sulfide generated by the reduction of dimethyl sulfoxide (a dimethyl sulfide smell was noticed during the reaction). The exact role of DMSO is not clear and is under investigation.
Table 1. [Enlarge]
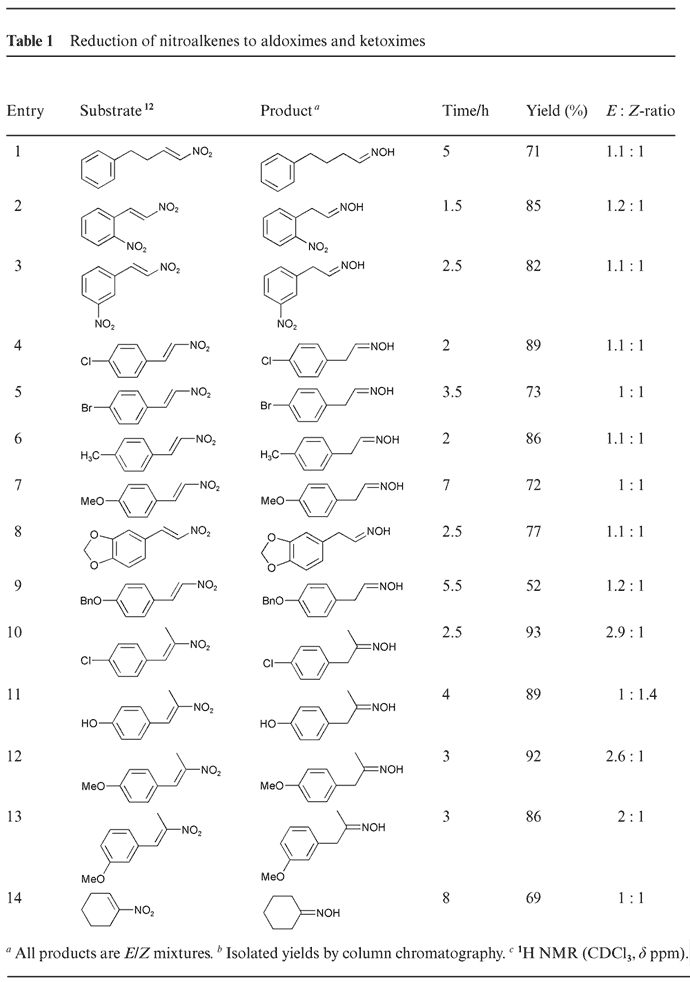
The reaction is chemoselective. An aromatic nitro group (entries 2 and 3) remained intact under our conditions.8 Even though dehalogenation is a known side reaction for catalytic transfer reductions of halogenated aromatic compounds2,3, it was not observed under our transfer hydrogenation conditions (entries 4, 5 and 10). The ratios of the E–Z mixture are listed in Table 1, which were determined using 1H NMR spectroscopy.
Side products such as ketones were not observed at all (entries 10–14). The reaction appears to be general as evidenced by the formation of phenylacetaldehyde oxime from β-nitrostyrene (entries 2–9), and cyclohexanone oxime from 1-nitro-1-cyclohexene, which are not obtainable under acidic conditions (entry 14).
In conclusion, the reduction of α,β-unsaturated nitroalkenes, using a system of decaborane and DMSO in methanol in the presence of 10% Pd/C at rt under nitrogen, generate the corresponding oximes in good to high yields. DMSO is necessary for the reactions to be efficient and the reactions are chemoselective. The reactions are mild11 and efficient.
Experimental
General procedure
Nitroalkene (50 mg) was stirred with decaborane (30 mol%), DMSO (5 eq.) and 10% Pd/C (30% w/w of nitroalkene)11 in methanol (20 ml) at rt under nitrogen. The reaction was monitored by TLC using a solution of ethyl acetate and hexane (1:4). After the reaction, the reaction mixture was filtered through a Celite bed and the filtrate was concentrated under reduced pressure. Saturated sodium bicarbonate solution (5 ml) was added to the concentrate and extracted with ether (3×5 ml). The ether layer was dried with anhydrous sodium sulfate and concentrated to give the corresponding oxime, which was sufficiently pure according to 1H NMR. For further purification, the concentrate was chromatographed on a short silica gel pad using a solution of 2% methanol in methylene chloride to give the corresponding oximes in good to high yields (Table 1).