Abstract
2-Methoxy-3,4-methylenedioxybenzaldehyde (croweacinaldehyde) of high purity was prepared in 55% overall yield from pyrogallol.
The title compound, 2-methoxy-3,4-methylenedioxybenzaldehyde (croweacinaldehyde, 1), was first obtained as an oxidative degradation product of 1-allyl-2-methoxy-3,4-methylenedioxybenzene (croweacin) occurring in the essential oil separated from the leaves and terminal branchlets of Eriostemon crowei1. At the same time, 1 was synthesized from 7,8-dihydroxycoumarin (daphnetin) to confirm the structure. This aldehyde 1 has been used as a key intermediate for syntheses of a variety of natural products2,3b Among several routes in the preparation of 11,3, formylation of 1-methoxy-2,3-methylenedioxybenzene (4) seems to be the most straightforward method although the isolated yield of 1 by recrystallization is relatively low because of the formation of the undesirable isomer 5. Compound 4 has been conveniently prepared by methylenation of 3-methoxypyrocatechol (3) which was derived from o-vanillin (2-hydroxy-3-methoxybenzaldehyde)4. But this method is not suitable for large-scale preparation because o-vanillin, which is usually obtained as a byproduct of the production of vanillin, is not easily available in quantity.
Pyrogallol (2) would be a good alternative to o-vanillin because it can be supplied in large quantities at reasonable cost. However, monomethylation of pyrogallol (2) has been reported to give 3 in very low yield4b, and preparation of 3 by methylation of 2,3-carbonyldioxyphenol with diazomethane and subsequent hydrolysis5 is also not suitable for large-scale production.
Since we needed a large quantity of 1 of high purity for the synthesis of 5,6,7,8-tetrahydro-4-methoxy-6- methyl-1,3-dioxolo[4,5-g]isoquinolin-5-ol (cotarnine)6, new processes to obtain 1 from pyrogallol (2) were investigated. Here, we report the efficient synthesis of 1 from pyrogallol (2). The synthesis involves recovery of 4 by decarbonylation of the undesirable isomer 5 formed by formylation.
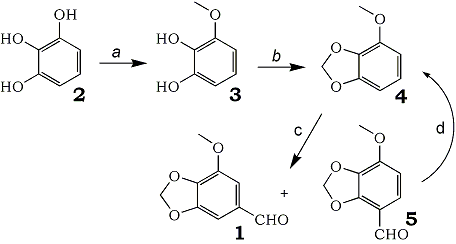
a) Me2SO4, NaOH, H3BO3, H2O
b) CH2Br2, NaOH, Bu4NBr
c) PhN(Me)CHO, POCl3 d) Pd-C.
Scheline7 reported the synthesis of 3,4-dihydroxy-5-methoxybenzoic acid from methyl gallate by the blocking of two vicinal hydroxyl groups in aqueous borax solution. He used a large excess of dimethyl sulfate as methylating reagent in large reaction volume. Through a modification of his procedure, pyrogallol (2) was selectively methylated with 1.5 equivalent dimethyl sulfate in boric acid solution. The pH of the solution was maintained at 10.0-10.4 by the addition of NaOH during the methylation. Yield of 3 was 84% (HPLC analysis), and 1,2,3-trimethoxybenzene, 2,3- and 2,6-dimethoxyphenol were detected as byproducts. Reaction condition out of this pH range required more dimethyl sulfate. The aqueous alkaline solution of crude 3 after the addition of NaOH was washed with CH2Cl2 to remove 1,2,3-trimethoxybenzene and then methylenated in a boiling mixture of CH2Br2, H2O, and Bu4NBr as a phase transfer catalyst8. Extracts of the reaction mixture were purified by distillation under reduced pressure to give pure 4 in 69% isolated yield from pyrogallol (2). Dimethoxyphenols might be methylenated in an intermolecular fashion9, and the resultant products could be removed by distillation.
Although there are many reports on the Vilsmeier formylation of 410, we have thoroughly reinvestigated the factors affecting the selectivity of formylation, because small changes of selectivity greatly affect the isolated yield of 1 by recrystallization. We found that the combination of N-methyl-formanilide and POCl3 was best for the formylation, and the optimal reaction temperature was 35-40°C. Under these reaction conditions, the formation ratio of 1 to 5 was 72 to 28. Reaction with other formylating reagents such as DMF-POCl3, or reaction at higher temperature resulted in a lower ratio. The reaction mixture was poured into ice water, and a mixture of 1 and 5 (1:5 = 74:26, 5 is more soluble than 1 in H2O) was obtained in total 93% yield. This mixture was recrystallized from 40% MeOH-H2O11 at 50°C to give 1 of 99% purity in 54% yield from 4. Solubility of 5 in 40% MeOH-H2O is greater than that of 1, and the solubility ratio of 5 to 1 at 50°C is greater than that at room temperature.
From the cooled mother liquor, the mixture of 1 and 5 (36:64) was recovered in 35% yield based on 4. This mixture was easily decarbonylated12 at 200°C over 5% Pd on carbon (0.5 mol%) to afford 4 in 92% yield, which would again be used for formylation.
In these processes a desired 1 of high enough purity for further use6 was prepared in 55% overall yield from pyrogallol (2) in combination with recovery of 4 from the mixture of 1 and 5 by decarbonylation.
Experimental
All reactions were carried out under a nitrogen atmosphere. HPLC was performed on a Shimadzu LC-6A liquid chromatograph equipped with a Nucleosil ODS column (4.6 mm x 25 cm) and a UV detector, and operated at a flow rate of 1 mL/min with a solvent composed of MeCN-H2O-AcOH (30:70:5) or MeOH-H2O (40:60).
3-Methoxypyrocatechol (3)
To a stirred solution of pyrogallol (2) (12.61 g, 0.1 mol) and boric acid (6.80 g, 0.11 mol) in 1 N NaOH (110 mL) was added dimethyl sulfate (4.72 mL, 0.05 mol) at 20-25°C, and the pH of the solution was maintained at 10.0-10.4 during the reaction by the addition of 25% NaOH. Two portions of dimethyl sulfate (2x4.72 mL, 0.1 mol) were added after 20 min and then 60 min, and the reaction mixture was stirred for another 2 h. In total 24 g (0.15 mol) of 25% NaOH was added. After another addition of 25% NaOH (32 g, 0.2 mol), the entire mixture was washed with CH2Cl2 (2x30 mL) to remove 1,2,3-trimethoxybenzene. The resultant aqueous alkaline solution of 3-methoxypyrocatechol (3) was used in the next step.
1-Methoxy-2,3-methylenedioxybenzene (4)
To a refluxing mixture of dibromomethane (34.76 g, 0.2 mol), tetrabutylammonium bromide (1.61 g, 0.005 mol), and H2O (20.7 mL) was added dropwise the alkaline solution of 3-methoxypyrocatechol (3) prepared above over a period of 4 h. The reaction mixture was further refluxed for 1 h, cooled, and extracted with CH2Cl2 (2x50 mL). The combined extracts were dried over anhydrous MgSO4, and concentrated. The residual oil was distilled under reduced pressure to give pure 1-methoxy-2,3-methylenedioxybenzene (4) (10.44 g, 69% yield from 1) as a clear oil which solidified readily; bp 120-122°C at 21 mmHg (lit.13 82°C/2 mmHg), mp 41°C (lit.4b 41°C).
2-Methoxy-3,4-methylenedioxybenzaldehyde (1)
To a mixture of 4 (15.22 g, 0.1 mol) and N-methylformanilide (20.28 g, 0.15 mol) was added dropwise POCl3 (13.98 mL, 0.15 mol) over a period of 20 min at 35-38°C. The mixture was stirred at 35-38°C for 5 h, then at 50°C for 1.5 h, and poured into ice water (236 g) with stirring. After the mixture had been further stirred for 1 h at 20-25°C, the deposited solid was collected by filtration, washed with H2O (2x74 mL), and dried at 40°C in vacuo to give 17.46g of an almost white solid, which contained 12.32 g of 1 and 4.42g of 5 (HPLC analysis, 93% combined yield). To a solution of the crude product (17.05 g) in MeOH (101 mL) was added dropwise H2O (150 mL) at 60°C, and then the mixture was stirred at 50°C for 2 h. The white crystal was collected by filtration, washed with 40% MeOH-H2O (20 mL) at 50°C, and dried in vacuo to afford 1 (9.61 g, 99% pure, 54% yield from 4) ; mp 103°C (lit. 104°C). The mother liquor was cooled to 5°C, and the deposited solid was collected by filtration and washed with cold 40% MeOH-H2O (40 mL). The product was dried to give the mixture of 4 and 5 as slightly yellow needles (6.23 g, 35% yield from 4, 1:5 = 36:64).
1-Methoxy-2,3-methylenedioxybenzene (4) by decarbonylation of the mixture of 1 and 5
The recovered mixture of 1 and 5 (5.41 g, 0.03 mol) with 5% Pd on carbon (0.32 g) was gradually heated to 200°C and stirred at this temperature for 2 h. After cooling, CH2Cl2 (30 mL) was added, and Pd on carbon was removed by filtration. The filtrate was concentrated and the residual oil was distilled under reduced pressure to give pure 4 (4.20 g, 92% Yield); bp 130°C/25 mmHg.