Procedure
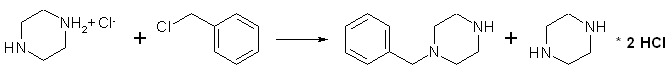
A solution of 24.3 g. (0.125 mole) of piperazine hexahydrate in 50 ml. of absolute ethanol, contained in a 250-ml Erlenmeyer flask, is warmed in a bath at 65°C as there is dissolved in the solution, by swirling, 22.1 g. (0.125 mole) of piperazine dihydrochloride monohydrate (Note 1). As warming in the bath at 65°C is continued, there is added during 5 minutes, with vigorous swirling or stirring, 15.8 g (14.3 ml, 0.125 mole) of recently distilled benzyl chloride. The separation of white needles comes almost immediately. After the solution has been stirred for an additional 25 minutes at 65°C, it is cooled, and the unstirred solution is kept in an ice bath for about 30 minutes. The crystals of piperazine dihydrochloride monohydrate are collected by suction filtration, washed with three 10ml portions of ice-cold absolute ethanol, and then dried. Recovery of the dihydrochloride is 21.5-22.0 g. (97-99%) (Note 2).
The combined filtrate and washings from the piperazine dihydrochloride are cooled in an ice bath and treated with 25 ml of absolute ethanol saturated at 0°C with dry hydrogen chloride (Note 3). After the solution has been well mixed, it is cooled for 10-15 minutes in an ice bath. The precipitated white plates of 1-benzylpiperazine dihydrochloride are collected by suction filtration, washed with dry benzene, and dried. The product, which melts at about 280°C with decomposition, after sintering at about 254°C (Note 4), amounts to 29-29.5g (93-95%). A solution of this salt in 50ml of water is made alkaline (pH > 12) with about 60 ml of 5N sodium hydroxide, then extracted twelve times with 20ml portions (Note 5) of chloroform. The combined extracts are dried over anhydrous sodium sulfate, and the pale brown oil (Note 6) remaining after removal of solvent is distilled at reduced pressure in a Claisen flask. The yield of pure 1-benzylpiperazine, bp 122-124°C/2.5 mmHg, is 14.3-16.5g (65-75%).
Notes
- Piperazine dihydrochloride monohydrate, which is recovered almost quantitatively in this procedure, may be readily prepared in essentially quantitative yield from the free base by the following procedure.
- A brisk stream of hydrogen chloride gas is passed for 5-8 minutes into a solution of 24.3g (0.125 mole) of piperazine hexahydrate in 50 ml of absolute ethanol contained in a 250-ml Erlenmeyer flask. A wide gas-inlet tube (about 10mm) is used to avoid clogging, and the flask is cooled in an ice bath to keep the temperature at about 25°C. After the gas stream has been discontinued, the contents of the flask are cooled to about 0°C, and the crystalline product is collected by suction filtration and washed with two 25ml portions of ice-cold absolute ethanol. The yield is about 22g (0.125 mole).
- If the filtrate from this isolation is evaporated to dryness at reduced pressure, crude 1-benzyl-4-piperazinium chloride is left as a residue. For removal of any piperazine dihydrochloride the chloride may be crystallized after rapidly filtering a hot solution in about 50 ml of absolute ethanol. Concentration of the filtrate, followed by cooling, gives 12.4 g (84%) of 1-benzyl-4-piperazinium chloride as prismatic plates, mp 167-168°C. This salt may be converted to the dihydrochloride by treatment with ethanolic hydrogen chloride.
- When absolute ethanol is saturated with hydrogen chloride at 0°C, the resultant solution is about 10.5N in hydrogen chloride.
- The melting point has been reported as 253°C by Baltzly, JACS 66, 263 (1944).
- The checkers found continuous extraction with chloroform to be convenient.
- The free base rapidly absorbs carbon dioxide on exposure to air and should therefore be protected during both manipulation and storage. The undistilled oil may be converted in good yield to 1-benzoyl-4-benzyl- piperazine hydrochloride, mp 245-245.5°C, by treatment with benzoyl chloride in benzene solution.