Summary
The synthesis of (R)-(-)- and (S)-(+)-2-amino-1-phenylpropane-3,3,3-d3 via a modification of a published procedure for the unlabelled compounds is described. The preparation of the (S)-enantiomer involved in the first step the reduction of D-phenylalanine to (R)-2-amino-3-phenyl-1-propanol-1,1-d2 with lithium aluminum deuteride. The reduction product was treated with p-toluenesulfonyl chloride to give (R)-3-phenyl-2-(4-toluenesulfamoyl)propyl-1,1-d2 4-toluenesulfonate, which was reduced with lithium aluminum deuteride to (S)-N-(1-methyl-d3-2-phenethyl)-4-toluenesulfonamide. The latter compound was cleaved to (S)-(+)-2-amino-1-phenylpropane-3,3,3-d3 using naphthalene anion radical. The overall yield in the sequence was 32.8%, the isotopic purity of the product was 99%, and the enantiomeric purity >99%. The methyl ester of the amino acid could also be used as starting material in the synthesis.
Introduction
In conjunction with a program to study the metabolism and pharmacokinetics of 1-phenyl-2-aminopropane (amphetamine) in humans and laboratory animals using combined gas chromatography-mass spectrometry (GC-MS) as analytical tool, we were in need of samples of (R)-(-)- and (S)-(+)-amphetamine labelled with deuterium in the α-methyl group1. This isotopically labelled variant of amphetamine has been shown, as its N-triflurooacetyl derivative, to have suitable massspectral properties for use in distribution and metabolism studies of amphetamine involving gems as analytical technique1-3. While a convenient synthesis of racemic 1 has been described4 we have found that chemical resolution5-7 of small amounts of amphetamine via the d-tartrate salt is inefficient and results in unacceptable losses of material. A stereospecific synthesis of the labelled enantiomers was therefore sought.
Nichols, et al. recently described8 an asymmetric synthesis of phenylisopropylamines capable of yielding enantiomerically pure amphetamine. However, their procedure was considered unsuitable for our purpose, since the starting material in the synthetic sequence is phenylacetone, in which the protons of the methyl group may exchange under a variety of conditions involved in its preparation and further reactions.
In 1951 Karrer and Ehrhardt described9 the conversion of the ethyl ester of (R)-(+)-phenylalanine to (S)-(+)-amphetamine in 5% overall yield. We have found that their procedure, with some modification, is an attractive method for the preparation of deuterium-labelled amphetamines (R)-1 and (S)-1.
Discussion
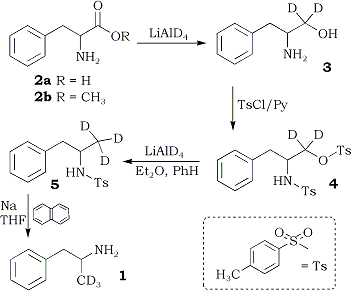
The sequence used for the preparation of the labelled pure enantiomers of amphetamine is shown in Scheme I. The reduction of (R)-phenylalanine ((R)-2a) to the labelled aminoalcohol (R)-3 was achieved directly10, without the intermediacy of an ester of the amino acid. Treatment of (R)-3 with p-toluenesulfonyl chloride gave (R)-3-phenyl-2-(4-toluenesulfamoyl)propyl-1,1-d2 4-toluenesulfonate ((R)-4) . The latter was reduced with lithium aluminum deuteride (LAD) to the labelled sulfonamide (S)-5. Conversion of (S)-5 to the desired labelled amphetamine (S)-1 via the method of Karrer and Ehrhardt9 appeared unattractive, since they obtained a 24% yield of impure amphetamine. It was recently reported that naphthalene radical anion cleaves sulfonamides to the parent amine in good yield11 and without racemization of an asymmetric center adjacent to nitrogen12. The reaction of this reagent with (S)-5 gave (S)-1, isolated as the recrystallized sulfate, in 50% yield.
The overall yield of (S)-1 was 32.8%. This compared well with the 39.4% yield obtained in the synthesis of racemic 14 and represents a significant improvement over the procedure of Karrer and Ehrhardt9. The methyl esters of (R)- and (S)-phenylalanine - both commercially available - may also be used as starting material in the synthesis.
The optical purity of (R)- and (S)-1 was determined to be at least 99% by gas chromatography of their N-(-)-α-methoxy-α-trifluoromethylphenylacetyl derivatives8,13. The isotopic purity of the labelled amphetamines was determined as 99% by GC-MS3.
The major advantages of the above synthetic route are the use of commercially available resolved phenylalanine as starting material, and the use of a convenient source of deuterium, lithium aluminum deuteride.
With the rapidly expanding applications of GC-MS in the biomedical sciences14-16 the use of stable-isotope-labelled compounds as internal standards, tracers, substrates for enzymatic reactions, etc., is also increasing17. We believe that the above-outlined procedure for the preparation of resolved labelled amphetamine will prove useful for the synthesis of other labelled compounds for which suitable amino acid precursors exist9.
Experimental
Melting points are uncorrected. L-Phenylalanine methyl ester hydrochloride and D-phenylalanine were obtained from the Sigma Chemical Company. Lithium aluminum deuteride was purchased from Merck, Sharp and Dohme Canada Ltd. Glass equipment used in reactions involving LAD was flame dried before use. Tetrahydrofuran (THF) was dried by distillation from lithium aluminum hydride immediately before use.
(R)-2-Amino-3-phenyl-1-propanol-1,1-d2 [(R)-3]
A 250-ml three-necked flask equipped with a nitrogen inlet line and a reflux condenser was charged with THF (80 ml) and LAD (4.1 g, 97.6 mmol). The contents of the flask were kept under a slight positive pressure of nitrogen, cooled in ice, and stirred magnetically while D-phenylalanine ((R)-2a) (6.5 g, 39.3 mmol) was added in small portions over 10 min. The resulting mixture was stirred at 0-5°C for 20 min and was then refluxed for 1.5 hr. The flask was cooled in an ice bath and ether (80 ml) was added, followed by cautious, dropwise addition of water (4.0 ml), 15% NaOH solution (4.0 ml), and water (12 ml). More ether (40 ml) was added, and the mixture filtered. The inorganic salts obtained were stirred with ether (100 ml) for 30 min and then filtered. The combined organic filtrates were dried (K2CO3) and evaporated to obtain 5.9 g of the aminoalcohol. Recrystallization from ether-petroleum ether gave 5.4 g (91.5%) of (R)-3, mp 86-8°C (lit9 mp 91.5°C).
(R)-3-Phenyl-2-(4-toluenesulfamoyl)propyl-1,1-d2 4-toluenesulfonate [(R)-4]
The aminoalcohol ((R)-3, 5.4 g, 35.1 mmol) was dissolved in dry pyridine (56 ml) and the solution was cooled in ice and stirred magnetically while p-toluenesulfonylchloride (17.7 g, 93.1 mmol) dissolved in pyridine (56 ml) was added dropwise. The resulting solution was stirred at room temperature for 25 hr. The reaction mixture was then poured into ice (300 g) and extracted with ethyl acetate (2x150 ml). The combined extracts were washed with 0.5 N HCl (3x500 ml) and water (2x200 ml), dried (Na2SO4), and evaporated. The residue, a brown oil, was dissolved in benzene (50 ml), charcoal was added, and the mixture boiled and filtered. The filtrate was treated with heptane until cloudy. The product crystallized in the cold overnight. Yield: 12.3 g (73.6%) of (R)-4, mp 94-6°C (lit9 98-98.5°C).
(S)-N-(1-methyl -d3-2-phenethyl)-4-toluenesulfonamide [(S)-5]
To a magnetically stirred slurry of LAD (4.0 g, 95.2 mmol) in dry ether was added dropwise a solution of (R)-4 (12.0 g, 26.0 mmol) in benzene (95 ml) over 30 min. The mixture was then refluxed, protected from moisture, for 24 hr. The flask was cooled in ice while water (4.0 ml), 15% NaOH (4.0 ml), and water (12.0 ml) were added cautiously, dropwise. The resulting mixture was stirred at room temperature, and filtered. The-inorganic salts obtained were dissolved in 1 N HCl (200 ml) and extracted with ether (2x100 ml). The ether extracts and the original filtrate were combined, dried (Na2SO4) and evaporated. A colorless oil (7.4 g, 97.4%) was obtained. The crude product was used in the next step without further purification.
(S)-2-Amino-1-phenylpropane-3,3,3,-d3 [(S)-1]
A solution of naphthalene anion radical was prepared as follows: naphthalene (14.0 g, 110 mmol) was dissolved in THF (225 ml) in a 500 ml Erlenmeyer flask equipped with a nitrogen inlet and outlet. The resulting solution was purged with nitrogen, and the reaction mixture was kept under nitrogen during subsequent operations. The solution was stirred magnetically using a glass-covered stirring bar while sodium (2.6 g, 111 mmol) cut into small pieces was added. The mixture was stirred at room temperature for 1 hr.
To the deep-green solution of the radical anion was then added a solution of (S)-5 (7.0 g, 24.0 mmol) dissolved in THF (20 ml) through the rubber septum serving as stopper, using a hypodermic syringe. The reaction mixture was stirred for 1 hr. Water was then added using the syringe until a colorless solution was obtained. The mixture was stirred until the residual small pieces of sodium had dissolved. The solution was then poured into water (200 ml) and extracted with ether (3 x 150 ml). The product amine was extracted from the ether solution into 1 N HCl (3x60 ml). The combined acidic solutions were cooled in ice, and solid NaOH (30.0 g) was added. The resulting basic solution was extracted with ether (3x60 ml), the ether extracts combined, dried (K2CO3) and evaporated. The residual oil (2.5 g) was dissolved in ether (25 ml) and was treated dropwise with conc. sulfuric acid (0.95 g, 9.3 mmol) dissolved in absolute ethanol (25 ml). The sulfate salt of (S)-1 , was collected (2.6 g, 58.9%) and recrystallized from ethanol-H2O-ether to give 2.2 g (50.0%) of (S)-1 sulfate.
(S)-2-Amino-3-phenyl-1-propanol-1,1-d2 [(S)-2]
(S)-Phenylalanine methyl ester ((S)-2b) hydrochloride (7.2 g, 33.4 mmol) was added to a solution of Na2CO3 (12.0 g) in H2O (100 ml) and the free base was extracted with ether (2 x 50 ml). The ether solution was dried (K2CO3) and evaporated. The residue (5.2 g) was dissolved in dry ether (50 ml) and added dropwise to a slurry of LAD (4.0 g, 95 mmol) in ether (80 ml). The mixture was stirred magnetically for 1.5 hr, followed by addition of H2O (3.9 ml), 15% NaOH (3.9 ml) and H2O (11.5 ml). The inorganic salts were filtered off and stirred with fresh ether (100 ml) for 30 min. The combined filtrate and ether washing were dried (K2CO3) and evaporated to yield 4.5 g (90.2%) of the aminoalcohol (S)-3.