Preparation of Optically pure Amphetamines by the way of alpha-Methylhydrocinnamic AcidJ. Org. Chem 22, 33-35 (1957)[ Back to the Chemistry Archive ] 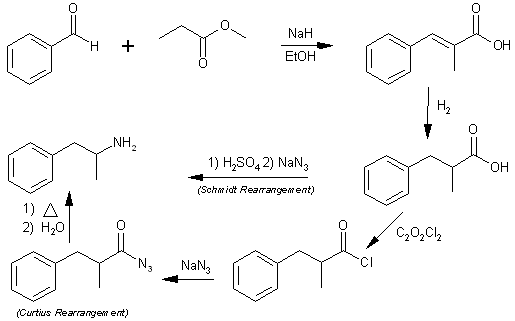
Abstract( - )-alpha-Methylhydrocinnamic acid (I), prepared by resolution with (+)-a-methylbenzylamine, was converted to optically pure ( - )-Amphetamine (II). An analogous series of reactions was carried out to form 3,4-dimethoxy-amphetamine. Experimentala-Methylcinnamic acid.A vigorously stirred suspension of 9.1 g. of sodium hydride in 125 ml. of methyl propionate was cooled in ice and treated with 0.45 ml. of absolute ethanol, then with 31.84 g. of benzaldehyde in 50 ml. of methyl propionate during 10 min. The ice bath was removed after one hour, replaced as soon as the vigorous reaction started, then removed again, and stirring was continued for another 2 hr. The organic layer was separated, after cautious addition of 28 ml. of acetic acid, then of 100 ml. of water, and the aqueous phase extracted with ethyl acetate. The combined ester solutions were washed with N hydrochloric acid and with sodium chloride solution, dried with sodium sulfate, and evaporated in vacuo. The residual ester was refluxed with 33.6 g. of potassium hydroxide in 60 ml. of water and 240 ml. of methanol for one hour. The solution was concentrated, diluted with water to ca. 500 ml., acidified with 6 N hydrochloric acid, chilled overnight, and the collected solid was washed with ice water; yield 35.67 g. (73%), m.p. 75-81°C'. The acid crystallized from hexane in colorless prisms, m.p. 80.5-81.0°C (lit[24] m.p. 81-82°C).3,4-Dimethoxy-a-methylcinnamic acid.The combined ester solutions, obtained from 18.53 g. of sodium hydride, 101.7 g. of veratraldehyde, and 356 ml. of methyl propionate, were washed with N hydrochloric acid, extracted with aqueous potassium carbonate, dried over potassium carbonate, concentrated, and passed through an alumina column which was then washed with chloroform. Evaporation of the eluates and crystallization from ether-hexane yielded 115.0 g. (80%) of the methyl ester as colorless prisms, m.p. 62.5-64.5°C (lit.[26] m.p. 65-67°C). The free acid was obtained in 99% yield by saponifying this ester with 2 molar equivalents of potassium hydroxide. It formed colorless needles, m.p. 142.8-143.8°C (lit. m.p. 142-143°C[25],144°C[26]). Additional acid, isolated by acidifying the potassium carbonate extract and by saponifying the evaporated mother liquors of the recrystallized ester, was purified by crystallization from ethanol, raising the total yield from veratraldehyde to 86%.DL-a-Methylhydrocinnamic acid (I)DL-a-Methylhydrocinnamic acid was prepared in 95% yield by treating 30 g. of a-methylcinnamic acid and 99.8 g.of sodium hydroxide in 900 ml. of water with 90 g. of Raney nickel-aluminum alloy at 90°C [27]. The cooled filtrate and washings were stirred into 720 ml. of concd. hydrochloric acid and about 700 g. of ice and extracted with ether (600 + 300 + 300ml). The ether extracts were washed with 2 N hydrochloric acid and with sodium chloride solution, dried, and evaporated. The distilled acid (b.p. 148-149°C/9 mm.,116.5°C/0.5 mm.) solidified on chilling; m.p. 36-37°C; yield 28.9 g. (95%). A sample crystallized from pentane in colorless prisms, m.p. 36.6-37.5°C (lit. [28] m.p. 37°C).DL-3,4-Dimethoxy-a-methylhydrocinnamic acid (IV)DL-3,4-Dimethoxy-a-methylhydrocinnamic acid was obtained in 94% yield as colorless hexagonal plates, m.p.61.5-62.8°C (lit. [29] m.p. 58-59°C), after evaporating the ether extracts and recrystalizing the residue from ether-pentane. L-(+)-a-Methylhydrocinnamic acid [10]A cold solution of 41.0 g . of racemic I in 350 ml. of ethyl acetate was treated salt. with 30.25 g. of ( - )-a-methylbenzylamine (III)[30], heated to dissolve the precipitate, and allowed to cool slowly. The solid was recrystallized six times from ethyl acetate to yield 16.1 g. of the (+)-I-( -)-III salt as long colorless needles, m.p. 127-129.5, [a]20/D -1.48" (c 2.00, chloroform). Systematic recrystallization of the second crops gave another 1.8 g. of pure salt, bringing the total yield to 50%.The free acid was obtained in 95% yield by shaking the salt with ether and excess 2 N sulfuric acid and extracting the aqueous phase31 with additional ether. The extracts were washed four times each with 2 Ar sulfuric acid and with sodium chloride solution, dried, and evaporated. Distillation gave (+)-I, b.p. 112°C/0.25 mm. D-( - )-a-Methylhydrocinnamic acid [10].The impure ( -)-I, recovered from the mother liquors of the (+)-I-( -)-III salt and treated with (+)-III[30], yielded 51% of the (-)-I-(+)-III salt, m.p. 127.3-129.5°C, which was also prepared from racemic I (after 10 recrystallizations: m.p. 128-130°C). The free acid had b.p. 111°C/0.22 mm.L-( + )-3,4-Dimethoxy-a-methylhydrocinnamic acid [10].The quinine salt was prepared from 96.7 g. of racemic IV and 140 g. of quinine in about 1800 ml. of ethanol and recrystallized thrice from the same solvent. It formed electrified needles, m.p. 163.7-164.3°C. The yield, including pure material isolated from the mother liquors, was 90%.The free acid was isolated in the same way as (+)- I; evaporation of the ethereal solution gave a viscous oil which, dried in a high vacuum, was used directly for further reactions. A sample was distilled in a bulb tube: b.p. 130°C (bath temperature)/0.01 mm. The acid chloride, prepared from 1 g. of the acid with 1.15 ml. of oxalyl chloride in 97% yield, had b.p. 90°C (bath temperature)/0.015 mm. The amide was obtained from the acid chloride with concentrated aqueous ammonia; it crystallized from water in colorless needles, m.p. 121.5-122.5°C. D-( - )-3,4-Dimethoxy-a-methylhydrocinnamic acid [10].Treating the impure (-)-IV, recovered from the quinine salt mother liquors, with (-)-III afforded the (-)-IV-( -)-III salt in 77% yield, based on the racemic IV used originally. The major portion was obtained pure after three recrystallizations from ethyl acetate; m.p. 107-109.3°C.The free acid had b.p. 120°C (bath temperature)/0.005 mm. The acid chloride had b.p. 95°C (bath temperature)/0.01mm. The acid chloride had b.p. 95°C (bath temperature)/0.01mm. D-( - )-a-Methylphenethylamine (Il) [10].( a ) By the Curtius Rearrangement. (-)-I (5.35 g.) was refluxed with 8.4 ml. of oxalyl chloride for one hour, and the solution evaporated in vacuo [32] and re-evaporated twice after adding dry benzene. The acid chloride, dissolved in 22 ml. of dry acetone, was added rapidly a -10°C to a magnetically stirred solution of 4.38 g. of sodium azide in 13 ml. of water. The mixture was stirred at 3°C for 1 hr. and extracted twice with a total of 37 ml. of benzene; the extracts were washed with sodium chloride solution, dried rapidly with sodium sulfate, filtered, and the residue was washed with another 15 ml. of benzene. The filtrate was concentrated at room temperature in vacuo [32] to about 1/2 of its volume, brought to about its original volume with dry benzene, heated to 62°C over 15 min. in the presence of a piece of Teflon (which seemed to promote nitrogen evolution), kept at this temperature for 1 hr., and evaporated in vacuo. The residue was warmed with 17.5 ml. of concd. hydrochloric acid at 45°C for 5 min., diluted with water, and the solution extracted once with ether to remove a slight opalescence. It was then treated with excess 10 N sodium hydroxide and extracted twice with ether. The extracts were washed with 10 N sodium hydroxide, dried with powdered sodium hydroxide, filtered, treated with 8 ml. of 5 N ethanolic hydrogen chloride, and evaporated to dryness. The residual ( - )-II hydrochloride was washed with ether and dried; yield 5.06 g. (90.5%), m.p. 155.7-156.7°C [18]. The neutral sulfate was obtained by treating the hydrochloride with 5 N sodium hydroxide, extracting the amine with benzene, and adding one equivalent of ethanolic sulfuric acid. It crystallized from water-ethanol-acetone in colorless leaflets [33].( b ) By the Schmidt rearrangement. A well-stirred solution of 7.02 g. of (-)-I in 50 ml of chloroform was treated with 13 ml. of concd. sulfuric acid, then at 45°C with 3.62 g. of sodium azide over 0.5 hr [20]. Stirring at 45°C was continued during 0.5 hr., and the mixture was worked up as under (a). The hydrochloride (yield 6.18 g., 84%) crystallized from ethanol-ether in colorless prisms, m.p. 153-155°C [18]. The neutral sulfate had m.p. 328-329°C (decompn. and darkening). D-( - )-3,4-Dimethoxy-a-methylphenethylamine (V) [10]Was obtained from (-)-IV by the Curtius rearrangement, following the procedure used for preparing (-)-II. The dried ethereal extract was evaporated, and the residue distilled (b.p. 96°C/0.08 mm.). The viscous oil (83% yield) solidified in the receiver; m.p. 37-40°C. A sample crystallized from pentane in long colorless needles, m.p. 39-40°CThe neutral sulfate was prepared in 91% yield with the exact amount of ethanolic sulfuric acid. It crystallized from dilute ethanol in colorless shiny leaflets, m.p. 313-315°C (decompn. and darkening). The hydrochloride was too deliquescent to be obtained pure. L-( + )-3,4-Dimethoxy-a-methylphenethylamineYield was 92%, had m.p. 37-40°C, and formed a neutral sulfate, m.p. 313°C (decompn.).Solubilities of a-methylbenzylamine salts (Table I).The pure salts (1 g.) were stirred magnetically in a glass-stop-pered Erlenmeyer flask at 35°C for 18 hr., then at 23°C for 1 hr , and the suspension was centrifuged. A 5 ml. aliquot of the supernatant was weighed in a stoppered weighing bottle and evaporated in an air current. The residue was weighed after drying in a vacuum desiccator.Table 1. Physical Constants of a-Methylbenzylamine Salts
a Determined in ethyl acetate at 23°C and expressed as grams/100g of solution. [10] The absolute D-configuration of (-)-I and (-)-III, shown in the formulas [11], follows from Karrer and Ehrhardt's [12] conversion of d-phenylalanine to L-(+)-II [13]. The D-configuration of (-)-IV and (-)-V has been demonstrated [14] by an analogous conversion of L-3,4-dihydroxyphenylalanine to N tosyl-(-)-V, which was found to have the same negative rotation as the compund obtained by the direct tosylation of (-)-V. [11] Cf. W. Klyne, Chemistry and Industry, 1022 (1951). [12] P. Karrer and K. Ehrhardt, Helv. Chim. Acta, 34, 2202 (1951). [13] The change from D to L in this conversion is due to the change in the chief function [14] A. W. Schrecker and J. L. Hartwell, to be published. This direct proof that (-)-I and (-)-IV belong to the same configurational series confirms the previously postulated [15] configuration of guaiaretic acid and related lignans. [15] A. W. Schrecker and J. L. Hartwell, J. Org. Chem., 21, 381 (1956). [18] The reported constants for (+)-II hydrochloride are: m.p. 147°C, 146°C, 156°C. [20] A. Campbell and J. Kenyon, J. Chem. Soc., 25 (1946). [24] R. Stoermer and G Voht, Ann., 409, 36 (1915). [25] A. Muller, M. Meszaros, K. Kormendy, and A. Kuszman, J. Org. Chem., 17, 787 (1952). [26] W.S. Ide and J. S. Buck, J. Am. Chem. Soc., 62, 425 (1940). [27] E. Schwenk, D. Papa, B. Whitman, and H. F. Ginsberg, J. Org. Chem., 9, 174 (1944). [28] M. Conrad and C. A. Bischoff, Ann., 204, 177 (1880). [29] R. D. Haworth, C. R. Mavin, and G. Sheldrick, J. Chem. Soc., 1423 (1934). [30] A. W. Ingersoll, Org. Syntheses, Coll. Vol 2. 506 (1943); W. Theilacker and H. G. Winkler, Ber., 87, 690 (1954). Both methods appear equally satisfactory. [32] A rotating evaporator was used. |