Abstract
Attempts have been made to synthesize 4-allylcatechol, the non-toxic antioxidant present in betel leaf. The catechol monoallyl ether on Claisen rearrangement gives both the 3- and 4-allylcatechols in 55 and 45% yields respectively. Photochemical rearrangement of catechol monoallyl ether in isopropanol solution under ultraviolet light for 120 hr gives 3- and 4-allylcatechols, each in 25% yield. In Claisen rearrangement, the anomalous behaviour, i.e. migration of allyl group to the para position when an ortho position is free, has been explained on the basis of the simultaneous progress of the reaction by both the normal ortho and the abnormal para migration.
In an earlier communication1 from this laboratory it was reported that hydroxychavicol (4-allylcatechol) isolated from betel leaf (Piper betle Linn.) is a powerful antioxidant for fats. Being present in a condiment which is eaten in India and other parts of Asia for centuries it has since long been considered as non-toxic and, therefore, ideal for use in edible fats. Moreover, amongst a number of catechol derivatives tested2 4-allylcatechol exhibited the maximum antioxidant activity. 4-Allylcatechol has been synthesized earlier3-5 by preparing the allyl ether of catechol and subjecting it to Claisen rearrangement. In all cases both mono- and diallyl ethers of catechol were formed. The catechol monoallyl ether on rearrangement gave 3- and 4-allylcatechols while the diallyl ether gave only 3,6-diallylcatechol. As the utility of 3-allylcatechol and 3,6-diallylcatechol as antioxidants for edible fats was limited due to their lower antioxidant index2 as well as their doubtful toxicity, it was considered worthwhile to explore alternative methods of synthesizing 4-allylcatechol in which no isomers are formed.
A modification of the earlier methods made it possible to completely avoid the formation of catechol diallyl ether. However, the catechol monoallyl ether on Claisen's rearrangement still yielded the 3- and 4-allylcatechols in 55 and 45% yields respectively.
It is well known that under Claisen rearrangement the allyl group migrates to an ortho position when even one unsubstituted ortho position is present, and to para position when both ortho positions are blocked. However, in the case of monoallyl ether of catechol, the allyl group migrates to the para position despite the availability of an unsubstituted ortho position. This anomalous behaviour, according to Tarbell6, is exhibited by derivatives of polyhydroxybenzenes. The ortho Claisen rearrangement occurs by an intramolecular cyclic mechanism.
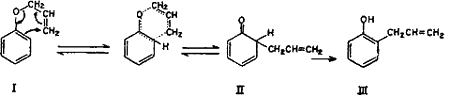
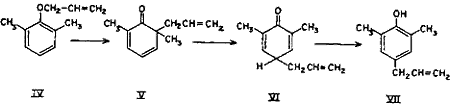
The unstable ketonic form of o-allylphenol (II) is produced as an intermediate, which spontaneously enolizes to the stable phenolic form (III)7,8.
The para Claisen rearrangement, which occurs when both ortho positions are substituted, begins in the same manner as migration to the ortho position and takes place in two stages.
The intermediate cyclohexadienone (V) cannot be stabilized by passing over to a phenolic system and hence the allyl group continues to move to the para position with the formation of a new cyclohexadienone (VI) which immediately changes over to the stable phenol (VII)9-14.
Though the ortho and para Claisen rearrangements have been studied so thoroughly, no explanation is available in literature for the anomalous behaviour of allyl ethers of mono-ortho-substituted polyhydroxybenzenes. The postulation (Chart 1) is an attempt to explain this anomaly. It can safely be presumed that a part of the rearrangement proceeds counter-clockwise by the ortho mechanism as for catechol monoallyl ether, thereby yielding the expected 3-allylcatechol (VIII).
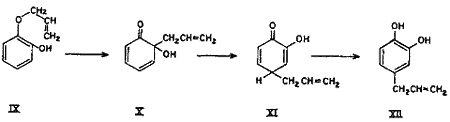
Simultaneously, under the activating influence of the ortho hydroxyl group, a parallel rearrangement (Chart 2) proceeds in the counter direction, i.e. by first forming an intermediate with the ortho hydroxyl group, thus giving rise to cyclohexadienone (X), which rearranges to the new cyclohexadienone intermediate (XI), and finally to 1,2-dihydroxy-4-allylbenzene (4-allylcatechol) (XII) as shown in equation. In other words, part of the rearrangement takes place by the expected ortho mechanism and the other part simultaneously by the para mechanism.
As this method of preparing 4-allylcatechol would always afford in addition the 3-allylcatechol, other alternative methods were tried.
Attempts to demethylate eugenol to obtain 4-allylcatechol in one step were made. The demethylating agents used were hydriodic acid, pyridine hydrochloride, anhydrous aluminium chloride, sulphuric acid and potassium hydroxide15. However, in each case a polymerized product was obtained.
Chart 1.
In the presence of boron trifluoride-acetic acid, guaiacol allyl ether rearranges at 68°C, and a para-allyl migration takes place, in spite of the fact that an ortho position is free16, some of the byproducts obtained being guaiacol, 6-allyleuginol allyl ether of allylguaiacol, etc.16. It was hoped that catechol allyl ether, when rearranged under these conditions, should yield 4-allylcatechol and the byproduct formation could be minimized by adding catechol to the system. Contrary to expectations, under these conditions, the byproduct formation happened to be the major reaction. Other boron trifluoride-catalysed reactions like condensation of allyl alcohol to catechol in various solvents, allyl bromide to catechol, etc., all failed because of polymerization.
Chart 2.
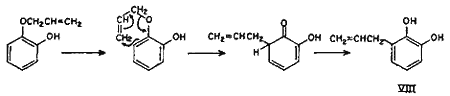
Kharasch et al.17 have pointed out that under the influence of ultraviolet light and in the presence of solvents like isopropanol, phenyl allyl ether and phenyl benzyl ether rearrange to give p-allylphenol and p-benzylphenol respectively, although under the thermal rearrangement (Claisen) only the ortho isomers are obtained. Schmid and Schmid18 have shown that in the photochemical rearrangements, the reaction proceeds by the dissociation and recombination mechanism. With a view to study the behaviour of catechol monoallyl ether under photochemical rearrangement conditions, it was dissolved in isopropanol and irradiated with ultraviolet light up to 120 hr. On working up and distillation, 4-allylcatechol in a maximum yield of 25% together with 3-allylcatechol in nearly the same amount (the rest being unreacted ether) were obtained. The gas-liquid chromatogram of the total reaction product did not reveal the presence of any other substance.
Polyphosphoric acid, an effective dehydrating agent, is also a mild condensing reagent. Generally it does not undergo a violet reaction with hydroxylic compounds19. Because of these properties it has been used for acylations of phenols20 and phenol ethers21. The acyl group goes preferentially to the para position to the hydroxyl group. Hence, it was decided to condense acrylic acid with catechol using polyphosphoric acid to get acryloyl catechol and then preferentially reduce the carbonyl group without affecting the double bond by using some of the newer reducing agents. Though acryloyl catechol was formed in 25% yield, all attempts to reduce it to 4-allylcatechol were unsuccessful.
Thus despite varied attempts to synthesize 4-allylcatechol exclusively, it has not been possible to obtain the required isomer only by any of the methods tried.
Experimental
Catechol monoallyl ether — To stirred catechol (44 g.; 0.4 mole), dry acetone (125 ml.) and freshly fused potassium carbonate (24.5 g.; 0.35 mole), allyl bromide (26 ml.; 0.3 mole) was added dropwise. The reaction mixture was refluxed for 4 hr, acetone recovered and the product cooled and filtered. From the residue consisting of unreacted catechol, potassium carbonate and potassium bromide, catechol (9.9 g.) was recovered by extraction with ether. The filtrate was dissolved in ether, acidified and washed free of acid. It was dried, concentrated and distilled when catechol monoallyl ether (40 g.; yield 88.9%); bp 80-82°C/0.8-1.2 mm.; n30D 1.5354 (lit.5 bp 103-4.5°C/8 mm.; n21.3D 1.5408) was obtained. No other fraction distilled over.
3- and 4-allylcatechols — Catechol monoallyl ether (30 g.; 0.2 mole) was heated in nitrogen atmosphere to 200-5°C (oil bath). A vigorous reaction ensued and the product changed colour to red. The temperature was maintained for 5 min. and then the product chilled in ice bath. Fractional distillation gave first fraction (13.5 g.) consisting of 3-allylcatechol; bp 110-12°C/3 mm.; n30(D) 1.5600 (lit.5 bp 132.8°C/9 mm.; n30(D) 1.5595) (Found: C, 71.8; H, 6.5; C9H10O2 requires C, 71.98; H, 6.71%). A second fraction (bp 120-3°C/2 mm.; n30(D) 1.5606; yield 11.8 g.) was also obtained. This was redistilled and crystallized from benzene-petroleum ether as colourless needles of 4-allylcatechol; mp 48-48.5°C (lit.5 mp 48.5°C; bp 141-4°C/7 mm.; n29(D) 1.5600). Both the allyl catechols gave deep green colour with ferric chloride solution.
Demethylation of eugenol — Eugenol was demethylated by the following methods: (i) Eugenol (10 ml.) petroleum ether (bp 100-20°C; 100 ml.) and aluminium chloride (15 g.) were refluxed for 3 hr and the reaction product worked up as usual. A colourless solid (mp 104-5°C) and a liquid (bp 108-15°C/0.3 mm.); n27.5(D) 1.5300) were obtained. (ii) Pyridine hydrochloride and eugenol in equal amounts were heated to 150°C for 3 hr, and the product worked up as usual. On distillation, a viscous liquid, bp 96-105°C/1 mm., was obtained (methoxyl content: found 18.3, required for eugenol 18.9%). (iii) Eugenol (10 g.), potassium hydroxide (30 g.) and absolute ethanol (325 ml.) were heated in an autoclave to 200°C for 8 hr. On working up a brown coloured polymerized product was obtained.
4-Allylcatechol by ultraviolet irradiation — Catechol monoallyl ether (30 g.) in isopropanol (50 ml.) was put in a quartz round-bottomed flask and irradiated for 120 hr by a 125 W. ultraviolet lamp in such a way that mild refluxing also took place. The product after the removal of the solvent was distilled to give the unreacted ether (14 g.), 3-allylcatechol (7.0 g.) and 4-allylcatechol (7.5 g.; 25% yield). The latter crystallized from benzene-petroleum ether in colourless needles; mp 48°C; n30(D) 1.5600; its dibenzoyl derivative, colourless plates; mp 72°C.
Gas-liquid chromatography on Griffin vapour phase chromatographic apparatus (MK-IIA) with katharometer as the detector did not show the presence of any other compound in the reaction mixture.
Synthesis of 4-allylcatechol using boron trifluoride as catalyst — (i) Allyl alcohol (11.6 g.; 0.2 mole) was saturated with boron trifluoride at 0°C. The complex formed (0.01 mole) was dissolved in dry ether (100 ml.) and catechol (0.01 mole) added. The mixture was then warmed and stirred for 2 hr. On working up a red coloured viscous mass was obtained, (ii) Boron trifluoride was passed into a solution of catechol (6.15 g.; 0.55 mole) and allyl alcohol (2.9 g.; 0.5 mole) in dry chloroform (100 ml.) at 0°C. After some time the reaction became too violent and the mass polymerized, (iii) Boron trifluoride was passed to saturation in a solution of diacetoxy catechol (2.1 g.; 0.011 mole), allyl alcohol (0.58 g.; 0.01 mole) and chloroform 50 mL at 0°C. The reaction was worked up after boiling in methanolic alkali for 30 min. A red coloured viscous liquid was obtained which on extraction and crystallization gave colourless needles; mp 120°C. This shows that 4-allylcatechol was not formed, (iv) Catechol monoallyl ether (15 g.; 0.1 mole), boron trifluoride-acetic acid complex (0.5 ml.) and catechol (0.5 g.) were heated to 95°C, with stirring, when an exothermic reaction took place and the internal temperature rose to 120°C, and the colour changed to red. On working up and distillation a colourless liquid (4.3 g.), bp 92°C/0.8 mm., was obtained; the rest of the mixture was a polymerized product.
Acryloyl catechol — In a flask, phosphorus pentoxide (45 g.) and phosphoric acid (20 ml.) were mixed to a homogeneous mass, heated to 110°C and catechol (5.5 g.; 0.05 mole) and acrylic acid (3.6 g.; 0.05 mole) were added to it all at once. After 10 min. of vigorous stirring, the reaction mixture was cooled and left overnight. Hydrochloric acid was added to it and the mass extracted with ether. The solvent layer was washed, dried and concentrated. The residue was extracted in boiling petroleum (60-80°C). The extract was concentrated and distilled to give a first fraction (2.1g; 25% yield); bp 105-10°C/1 mm.; nD30 1.5368 and a second fraction; bp 160-70°C/1 mm.; nD30 1.4680. The first fraction showed unsaturation, and gave a 2,4-dinitrophenylhydrazone; mp 189°C.
All attempts to reduce the carbonyl group without affecting the adjacent double bond to give 4-allylcatechol were unsuccessful.