Synthesis of 4-Alkylthio-2,5-DimethoxybenzaldehydesBy B.L. Cassels and S. Sepulveda-Boza, Bol. Soc. Chil. Quim. 35, 313-316 (1990)Typing and Spanish translation by Sunlight, HTML and pictures by Rhodium [ Back to the Chemistry Archive ] Abstract2,5-Dimethoxybenzaldehyde was brominated in acetic acid to give 4-bromo-2,5-dimethoxy- and 2-bromo-3,6- dimethoxybenzaldehyde in 4:1 ratio. The former derivative underwent nucleophilic substitution with thiols in DMF at room temperature in the presence of K2CO3 to afford the corresponding 4-alkylthio-2,5-dimethoxy- benzaldehydes in excellent yields. IntroductionIn the course of an investigation about the effect of the C-4 lipophilic substituent on the pharmacological characterisitcs of the 2-(2,5 dimethoxy-4-X-phenyl)-1-aminoethanes and 1-(2,5 dimethoxy-4-X-phenyl)-2 aminopropanes was necessary to have a series of 4-alkylthio and 4-(2-Y-alkylthio)- 2,5-dimethoxybenz- aldehydes as synthetic intermediates. The 4-methylthio-2,5-dimethoxybenzaldehyde derivative had been prepared for more than a decade ago by chlorosulfonation of 1,4-dimethoxybenzene, reduction of the formed 2,5-dimethoxysulfonyl chloride, S-methylation of the free thiol and Vilsmeier formylation [1] and other higher homologs have been synthetized through a similar route, though for (2) there aren't any experimental details available [2]. More recently a preparation [3] was described of 2,5-dimethoxy-4-methyl- thiobenzaldehyde and its 4-benzylthio analog by nucleophilic substitution of 4-bromo-2,5-dimethoxybenzaldehyde in DMF with the respective sodium salts of methyl and benzyl mercaptane, This synthesis involves the preparation of the sodium mercaptides with NaH under nitrogen. An easier way, with similar mechanistics to this last one seemed initially viable, general, and considerably shorter. It is known that the displacement of an aromatic nitro group in a ortho-nitrobenzaldehyde by methyl thioglycolate proceeds in an acceptable yield at room temperature in less than 24 hours when using DMF as solvent [4]. A similar activation of a para-nitro group in regards to a carbonyl function, although substituted by alkoxyl anions, was previously communicated [5]. According to an investigation in 1925, nitration of commercially available 2,5-dimethoxybenzaldehyde with concentrated nitric acid [d 1.42] would result in 3,6-dimethoxy-2-nitrobenzaldehyde (2) and 2,5-dimethoxy-3-nitrobenzaldehyde in a 4:1 ratio [6]. Nevertheless, it has been pointed that at -20°C the minor product would really be 2,5-dimethoxy-4-nitro- benzaldehyde (1) [7]. In our own hands, working at room temp, this result was repeated confirming thus the structure of the products from their 1H NMR. The 2,5-dimethoxy-3-nitrobenzaldehyde (1) necessary for subsequent reactions is formed in only a small proportion and an efficient purification of the compound can only be gotten by column chromatography, consequently it is better to use another substrate for nucleophilic substitution. Taking into account that bromide ion is generally a nucleophobe less reactive than nitrite ion, we consider the possibility of working with 4-bromo-2,5-dimethoxybenzaldehyde (3) if it could be easily accessible. Bromination of 2,5-dimethoxybenzaldehyde was originally described together with its nitration 65 years ago [6], stating that the electrophilic attack is mainly at the ortho position in regard to the formyl group to give 2-Bromo-3,6-dimethoxybenzaldehyde (4). This conclusion has been recently taken up and rationalized again [8], but a recently published hard-to-access article [9] points out that the product of reaction is the 4-bromo-2,5-dimethoxylated isomer (3). We have always obtained a mixture of both benzaldehydes, dominated the 4-bromo derivative (3), accompanied with approximately a 20% of the 3-bromo derivative (4). By this method 4-bromo-2,5-dimethoxybenzaldehyde can easily be obtained from 2,5-dimethoxybenzaldehyde, and is easily and quickly purified in good yield by simple recrystallization. Recent scientific literature mention the preparation of this aldehyde by bromination of 2,5-dimethoxybenzaldehyde in DCM in the presence of SnCl4 [10], a route that at this present moment wouldn't offer any advantage. 2-Bromo-3,6-dimethoxybenzaldehyde (4) is also easily isolated from the bromination mother liquor, so catalyst-free bromination can be considered perfectly accurate to obtain both isomers. Following the reaction model of displacement of the nitro group of ortho-nitrobenzaldehydes by methyl thioglycolate [4], we found that the substitution of halogen atom of 4-bromo-2,5-dimethoxybenzaldehyde using thiols as nucleophiles in DMF and in the presence of K2CO3 takes place at room temp and it is practically complete in 48 hours. In this way we obtained the 4-ethylthio- (5), 4-propylthio- (6) and 4-(2-hydroxyethyl)- thio-2,5-dimethoxybenzaldehydes (7) with excellent yields. Scheme 1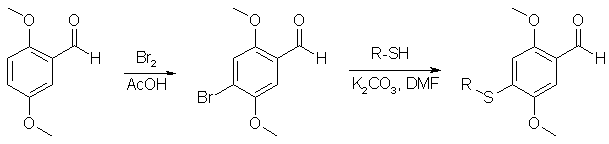
These compounds (Table 1) are described by the first time in the present communication.
Experimental Part2,5-Dimethoxy-4-nitrobenzaldehyde (1) 2,5-dimethoxybenzaldehyde (3.00 g, 18 mmol) diluted in AcOH (5mL) was treated with 12 mL 65% HNO3 in small portions, with good stirring and cooling in a water bath. The reaction mixture, partially crystallized, was left at room temp around 10 min and then it was diluted in 50 mL water. Filtering, washing with water and drying gave 3.27g (86%) of product, the majority being 3,6-dimethoxy-2-nitrobenzaldehyde (2), 4-nitro- 2,5-dimethoxybenzaldehyde (1) in ~20% yield, and a small quantity of a third unidentified isomer. The crude product was fractionated by chromatography in a silica gel column with ethyl acetate as eluent, from which firstly the minor product (1) appeared, which after recrystallization from IPA had a mp of 163-164°C (lit. 163-165°C [7]). 3,6-Dimethoxy-2-nitrobenzaldehyde (2) Continuing the elution of the column above it was obtained the main product (2). Recrystallization from IPA gave mp 163-164°C (lit. 164-165°C [7]). 4-Br-2,5-dimethoxybenaldehyde (3) 2,5-dimethoxybenzaldehyde (25 gr, 0.15 mol) was dissolved in AcOH (100 mL). A solution of Br2 (7.8 mL) was added in AcOH (48 mL) with good stirring and the reaction mixture were left to stand at room temp for 48 hours. After this time it was poured with vigorous stirring in cold water (750 mL), which caused an immediate crystallization of the solution. Subsequent filtration, washing with water and air drying gave 29g (79%) of a solid which 1H-NMR showed was mainly a mixture of 4-bromo-2,5-dimethoxy-benzaldehyde (3) and 2-bromo- 3,6-dimethoxybenzaldehyde (4) with an approximated ratio of 4:1. The main product was purified by recrystallization in MeOH, yielding 19.3g (53%) of pale yellow rectangular plates with sky blue fluorescence under a radiation of 366 nm, which sublimated to needles at 110°C, mp 130-134°C (lit. 132-133°C [10]). Chromatography of the concentrated mother liquors in a silica gel column (Eluent: hexane:DCM, 1:1), gave a small aditional amount of compound (3) in the first fractions (1.6 g, total yield 57%). 2-Br-3,6-dimethoxybenzaldehyde (4) Following with the elution of the chromatographic column described above it was obtained 2-bromo-3,6- dimethoxybenzaldehyde (4.7g, 13%), which could be recrystallized from i-PrOH to give needles and plates, which turned into needles at 95°C, mp 102-104°C (lit 125-126°C [6]). 2,5-dimethoxy-alkylthiobenzaldehydes (5), (6) and (7) 4-Bromo-2,5-dimethoxybenzaldehyde (18.1g, 74 mmol) was dissolved in DMF (185 ml) and was stirred 48 hours with powdered K2CO3 (10.2g, 74 mmol) and the respective thiol (148 mmol). The reaction mixture was poured onto water and ice (2 L) with good stirring and the precipitated solid was filtered, washed with water and dried, and then recrystallized in MeOH or in MeOH-water. Yields and mp of these products are in Table 1. References [1] P. Jacob, III, A.T. Shulgin and N. Castagnoli, Jr., J.Med. Chem. 20, 1235 (1977)
|
|||||||||||||||