A new route to 3,5-dimethoxy-4-something-phenethylaminesby Labrat ProcedureNow this method is a real breakthrough in the field of organic chemistry. This little procedure makes it possible to make 3,5-dimethoxy-4-something-amphetamines and beta-phenethylamines. PIHKAL is filled with compounds having a 2,4,5-trisubstitution pattern, but with this protocol a chemist can make analogs of most of those compounds. It's time for PIHKAL II!!! The beauty of this reaction is that it uses the highly useful trimethoxybenzaldehyde. A synthesis of this compound is given earlier in this book. The procedure can be found in these useful articles [JOC 57,3101-6 (1992) and Synthesis 313 (1990)]. The reaction diagram is as follows:
1) Preparation of dimethylacetal of 3,4,5-trimethoxybenzaldehyde 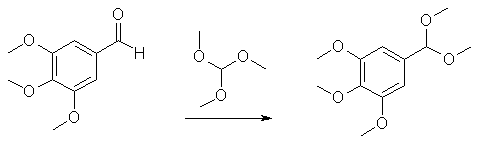
10 g 3,4,5-trimethoxybenzaldehyde is added under N2 to a suspension of 150 mg NH4Cl in a solution of 20 ml trimethoxymethane and 20 ml MeOH. The mixture is stirred at reflux for 3 hrs, then cooled to room temperature and 1.5 ml triethylamine is added with stirring. After a few minutes, 50 ml water is added and the mixture is extracted with 30 ml ether (3 times). The organic phase is washed with 30 ml saturated sodium carbonate solution, 30 ml water and dried with Na2SO4. After evaporation of the solvent a crude product remains, which is distilled at 180°C/10 mmHg to give 10.3 g 3,4,5-trimethoxybenzaldehyde dimethylacetal. This particular reaction makes a protected group from the aldehyde: an acetal. The function of this protection group is to protect the aldehyde group against the harsh reducing conditions in the second reaction. Otherwise the aldehyde would be reduced to an alcohol or a methyl group. 2) Reductive electrophilic substitution of the above acetal: 3,5-dimethoxy-4-methyl-benzaldehyde 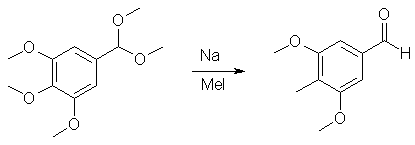
A solution of 4 g 3,4,5-trimethoxybenzaldehyde dimethylacetal in THF is added dropwise to a mixture of freshly cut Na (1.2 g) in 60 ml THF, cooled to 0°C under dry nitrogen. The mixture is stirred at room temperature for 24 hrs, cooled to 0°C and 25 mmol methyl iodide is added dropwise. After another 24 hrs of stirring at r.t. the reaction mixture is carefully quenched with 20 ml water, 30 ml ether is added and the organic phase is washed with 20 ml water and evaporated. The crude product is dissolved in 50 ml of a 1:1 mixture of THF/1 M HCl and stirred at room temperature for 5 hours. The mixture is extracted with 3x30 ml ether, the ether washed with 2x20 ml water and dried with Na2SO4. After evaporation the crude product is recrystallised from boiling acetone to give 71% yield of 3,5-dimethoxy-4-methyl-benzaldehyde. This reaction is actually two steps in one procedure. First, the trimethoxycompound is reduced with sodium to the aryl anion, then methyl iodide (an electrophile don't ask, I'll give examples of this later) is added to the mix producing the 2-Me compound in ONE step! Well, isn't that nice? A competing reaction is demethylation, which produces phenols. These latter substances are easily removed during workup (the phenols can be extracted into acid water). Those phenols should be saved to make Tweetios or analogue compounds. Labrat just mentioned something about electrophiles. The following electrophiles can be used to make 3,4,5-analogs of the following compounds:
Of course there are more example of electrophiles, but this would take too much of Strike space in the book. Included are the most interesting compounds. Labrat has added some details for the curious chemist:
The reaction is used to make Desoxy. In PIHKAL a difficult procedure with low yields overall is used to synthesize this particular compound, but this protocol is a much better way to do the same thing. Desoxy is synthesized from the 3,5-dimethoxy-4-methylbenzaldehyde as done in PIHKAL, #96: Mescaline. 3) 3,5-dimethoxy-4-methylnitrostyrene 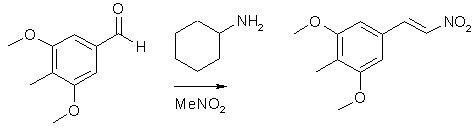
20 g 3,5-dimethoxy-4-methylbenzaldehyde, 40 ml nitromethane and 20 ml cyclohexylamine in 200 ml glacial acetic acid is heated at 100°C for 1 hour. The reaction mixture is slowly diluted with 400 ml water under vigorous stirring, giving a heavy precipitate of the nitrostyrene. The yellow crystals are filtered off, washed with water and dried. Then recrystallise from ~15 ml MeOH/g nitrostyrene to get about 20 g of the nitrostyrene. 4) Desoxy: 3,5-dimethoxy-4-methylphenethylamine 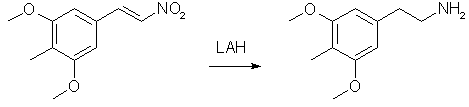
2 g LiAlH4 in 100 ml ether is brought to reflux and there's added dropwise a saturated solution of 2 g 2,5-dimethoxy-4-methylnitrostyrene in ether. After the addition the mixture was refluxed for 48 hours, then cooled to r.t. Excess hydride was decomposed with 150 ml 10% H2SO4. Separate the two layers that form in a sep funnel, keeping the aqueous layer. Bring the pH to >9 with saturated NaOH solution and extract with 3X100 ml toluene. Bubble dry HCl gas through the solution until no more crystals precipitate. Filtering is done when necessary. Yield ~2 g of Desoxy. |