Abstract
3-Hydroxythiophenol is a versatile intermediate for the synthesis of medicinal and heterocyclic compounds. Two novel and practical syntheses of 3-hydroxythiophenol are achieved using readily available and inexpensive starting materials. An overall cost comparison of these syntheses is also given.
Hydroxythiophenols are important starting materials for a variety of medicinal and heterocyclic compounds.2 The 3-hydroxythiophenol moiety, in particular, is present in the osteoporosis drug, raloxifene.2a An early synthesis of 3-hydroxythiophenol (3-HTP) involved hydrolysis of monothioresorcinol-O-carboxylic acid ethyl ester which was made from 3-hydroxybenzenesulfonic acid sodium salt in three steps.3 A later procedure included reaction of the diazonium salt of 3-aminophenol with aqueous potassium ethyl xanthate at 70°C.4 However, diazonium xanthates (ArN = NSCSOC2H5) can be explosive and violent reactions have been reported.2 The 3-hydroxythiophenol was also obtained when 3-methoxyphenyl methyl sulfide was treated with two equivalents of sodium diethylamide and hexamethylphosphoric triamide in refluxing xylene.2 Reaction of hydrogen sulfide with 3-chlorophenol at high temperature (500-600°C) has also been reported to yield 3-hydroxythiophenol.7 Recently, we had a need to produce commercial quantities of 3-HTP in high purity. These existing literature preparations are unsatisfactory as a viable basis for a scaleable process because of potential safety hazards or because the required starting materials are not commercially available.
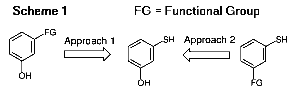
At the outset, we envisioned two approaches to investigate (Scheme [1]). The first approach is to start from a precursor having a phenolic hydroxyl functionality already in place and a second functional group that can be readily transformed into an aryl thiol group. A survey of the literature revealed two potential thiol-forming functional groups, namely, arylamines and aryl halides. Thearylamines can be converted to the corresponding aryl thiols via their diazonium salts. Nucleophilic displacement of the diazonium group with a sulfur nucleophile readily yields the thiol.4 The synthetic utilities of these reactions, however, are limited only to small-scale runs because of explosion risks.5 Aryl halides, on the other hand, can be converted to aryl thiols via reaction of their Grignard reagents with sulfur.8 To the best of our knowledge, there have been no reports in the literature on any potential explosion risks. Thus, halogenated phenols such as 3-bromophenol are "ideal" hydroxyl containing precursors for this purpose. The second approach is to start from a precursor containing an aryl thiol group and a second functional group that can be converted readily to a phenolic hydroxyl function. Since phenols can be readily made from aromatic amines via their diazonium salts, 3-aminothiophenol is an "ideal" thiol-containing precursor to 3-hydroxythiophenol. We wish to report herein two practical syntheses of 3-hydroxythiophenol based on the two aforementioned approaches.
The Grignard Reaction Approach
We investigated this approach first because of its high probability of success in the near term (Scheme [2]). 3-Bromophenol (1) was first protected as the trimethylsilyl ether 2 in high yield prior to the Grignard reaction. The initiation went very smoothly in the temperature range of 25-30°C in THF. Some decomposition of the Grignard reagent was observed, especially at elevated temperatures.9 The internal temperature was thus kept below 55°C during the bromide addition to minimize decomposition. Inverse addition of the Grignard reagent to a sulfur slurry in THF at 0-15°C led to the formation of the ArSMgBr intermediate. Quenching with dilute aqueous HCl removed the TMS protective group to afford the desired 3-hydroxythiolphenol. Fractional vacuum distillation yielded the desired compound in >99% purity (GC).
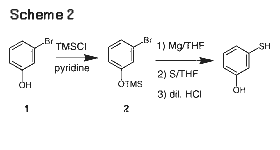
The choice of the protective groups is critical to the reaction. Initially we explored the tetrahydropyranyl (THP) group, a typical hydroxy protective group for Grignard reaction. While the Grignard reaction went very smoothly in high yield, the subsequent reaction with sulfur produced low and variable yields (0-25%) of the desired hydroxythiophenol. Examination of the major by-product revealed the reactive nature of the SH group toward dihydropyran, formed in the HCl quenching/de-protection step. A silyl protective group is advantageous in this particular case because of the low reactivity of TMSOH toward the SH group.
The Grignard route to 3-hydroxythiophenol was scaled up in a production plant. A total of 11 batches (ca. 100 kg/batch) were produced in consistent yields and purity. This route supplied us material for the near term, however, it is not cost-effective as a production process. The key raw material 3-bromophenol was expensive and suffered from poor weight throughput due to replacement of the bromine atom with mercaptan group. Furthermore, multiple labor-intensive extractive work-up and distillation steps were required. We thus explored the second approach as elucidated earlier.
The Diazonium Salt Approach
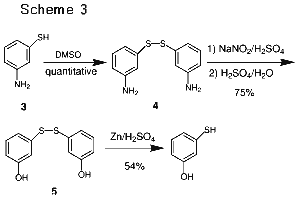
The success of this approach is hinged on the protection of the thiol group prior to the diazonium salt formation, to avoid its potentially hazardous reaction with the diazonium salt. Since cost reduction was the main driver for the alternate process, the selected protective group(s) had to meet the requirements of good atom economy and low cost. Internal protection via disulfide formation best meets these requirements.
As shown in Schemes [3], 3-aminothiophenol (3) was first internally protected as the disulfide 4 in near quantitative yield via oxidation by methyl sulfoxide (DMSO). The disulfide 4 was then doubly diazotized followed by hydrolysis to afford the 3-hydroxyphenyldisulfide 5.10 Reduction of 5 with Zinc/sulfuric acid then led to the formation of the desired 3-hydroxythiophenol.
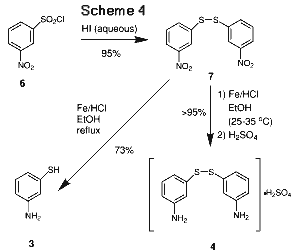
The 3-aminothiophenol (3) was initially purchased from commercial sources in small quantities. Its high cost, however, would prevent its use on commercial scale as the key starting material. Therefore, a cost-effective in-house synthesis of 3 was developed from 3-nitrobenzenesulfonyl chloride (6), an inexpensive commercial material. At first, we explored simultaneous reductions of both the nitro and sulfonyl chloride groups under various conditions. Using zinc/HCl in aqueous ethanol at reflux, 3-nitrobenzenesulfonyl chloride (6) was reduced to 3-aminophenyl disulfide (4) as its hydrochloride salt in low yields. A trace amount of 3-aminophenyl sulfonic acid ethyl ester was detected by GCMS. The majority of the product appeared to be metanilic acid, a compound from the hydrolysis of the sulfonyl chloride under the reaction conditions. Attempts to minimize the hydrolysis using 70% sulfuric acid did not give much improvement in yield. A stepwise approach was then pursued (Scheme [4] ). Reduction of the sulfonyl chloride with hydriodic acid afforded 3-nitrophenyl disulfide (7) in better than 95% yields, as reported in the literature.11 The nitro group was then reduced to NH2 using Fe in ethanolic HCl. Initially, the reduction was carried out under reflux conditions. Under these conditions, both the nitro and disulfide linkage were reduced yielding the desired 3-aminothiophenol (3) in moderate yields. To our pleasant surprise, the reduction became highly selective at lower temperatures (25-35°C) to give 3-aminophenyl disulfide (4) directly in near quantitative yield which was conveniently isolated as the sulfate salt. The sulfate was diazotized directly with no further purification and then converted directly to 3-HTP as described previously.
In summary, we have developed practical and scaleable syntheses of 3-hydroxythiophenol from either 3-bromophenol via the Grignard reagent or 3-nitrobenzenesulfonyl chloride via the diazonium intermediate. While both routes produce 3-hydroxythiophenol in good yields and high purity, the latter approach requires only inexpensive raw materials and provides convenient isolation of solid intermediates, thus offering major cost advantages.
Experimental:
3-Bromophenyl Trimethylsilyl Ether (2)
A dry, nitrogen purged 5 L 3-necked round-bottomed flask was equipped with a mechanical stirrer, a pressure-equalizing addition funnel and a Claisen adapter through which were fitted a reflux condenser and a thermocouple. To this flask was added THF (2340 mL), pyridine (294.5 mL, 3.6 mol) and 3-bromophenol (486.8 g, 2.8 mol) to obtain a homogeneous soln. To this soln was then added carefully, via the addition funnel, trimethylchlorosilane (464.8 mL, 3.6 mol) under nitrogen, with stirring in ca. 45 min while maintaining pot temperature below 50°C. The resultant white slurry was stirred at ambient temperature until the reaction was complete as shown by GC. The solids were removed by vacuum filtration using a Buchner funnel and washed with THF (2x200 mL). The filtrate was concentrated under reduced pressure to obtain the crude product as a liquid. The crude product was purified by distillation to afford 3-trimethylsiloxylbromobenzene (642 g, 94%) as a clear liquid; bp 77°C/0.4 mmHg.
3-Trimethylsiloxylphenylmagnesium Bromide
A dry, nitrogen purged 5 L 3-necked round-bottomed flask was equipped with a mechanical stirrer, a pressure-equalizing addition funnel and a Claisen adapter through which were fitted a reflux condenser and a thermocouple. The flask was charged with magnesium turnings (55.1 g, 2.26 mol), then heated to 110°C under nitrogen and cooled slowly to ca. 85°C at which point, iodine (400 mg) was quickly introduced. The flask was further cooled to below 40°C and THF (1224 mL) was added. To the stirred Mg/THF suspension, a portion (40 mL) of 3-trimethylsiloxylbromobenzene (500 g, 386 mL, 2.04 mol) was added via the addition funnel. The pot temperature was then raised to 36°C and the Grignard reaction initiated. The remaining bromide was then added carefully so that the reaction could be kept at 46-50°C. The addition funnel was rinsed with THF (117 mL) and the rinse soln was added to the reaction mixture. After the reaction was complete as shown by GC, an aliquot of the Grignard reagent was titrated by a literature procedure (1.55 mol, 76% yield). [9] The Grignard soln was held at 40°C before use to avoid crystallization.
3-Hydroxythiophenol
A dry, nitrogen purged 12 L, 4-necked jacketed flask equipped with a mechanical stirrer, a pressure-equalizing addition funnel, a reflux condenser and a thermocouple was charged with sulfur powder (52.1 g, 1.6 mol) and THF (585 mL). The suspension was cooled to 5°C with stirring. To this mixture was then carefully added the above Grignard reagent via a Teflon transfer tube by applying nitrogen pressure while maintaining the reaction temperature below 15°C. The flask containing the Grignard reagent was rinsed with THF (2x234 mL) and the rinse solns were also added to the reaction mixture. The reaction was warmed to r.t. and stirred until a negative Gilman’s test [12] is obtained. To the resultant mixture was then added 10% aq HCl (1158 g) in one portion. The reaction mixture was stirred for one additional hour while cooling to r.t. The bottom aq layer was drained off and the top THF layer was treated with a soln of KOH (508 g) and Na2S2O5 (212.7 g) in H2O (1740 mL) at reflux for 2 h. The THF layer was separated and concentrated under reduced pressure. The residue was combined with the aq layer and the combined aq phase was extracted with t-butyl methyl ether (2x1170 mL). The aq phase was acidified to pH 1 with concd HCl, and extracted with t-butyl methyl ether three times (1560, 1170, and 780 mL respectively). The combined organic layer was washed with brine (780 mL). Removal of the solvent under reduced pressure while keeping the water bath temperature <30°C afforded 181.6 g of the crude product. The crude was purified by fractional vacuum distillation using a Vigreux column to provide 3-hydroxythiophenol (135.7 g, 53%) as a colorless liquid; bp 92°C/0.075 mmHg.
Bis(3-nitrophenyl) Disulfide (7)
This procedure was adapted from Sheppard [11] with some modifications. A 500-mL, 3-necked, round-bottomed flask was equipped with a mechanical stirrer, a thermal couple and an addition funnel. The flask was charged with 3-nitrobenzenesulfonyl chloride (6) (50 g, 0.226 mol) followed by a rapid addition of hydriodic acid (47-49%, 306.7 g, 1.15 mol) in ca. 2 min while stirring. The addition funnel was replaced by a condenser. The reaction temperature slowly rose to 41°C in ca. 10 min and the mixture turned purple. The reaction was stirred at r.t. for 3 h followed by addition of an aq sodium bisulfite soln (73.4 g in 170 mL of H2O). The reaction mixture turned yellow. The precipitate was collected by vacuum filtration, washed with H2O (2x50 mL) and dried by suction. The solid was further dried in a vacuum oven at 50°C for 17 h to obtain 35.5 g of the crude product (102%). The crude was of sufficient purity and used for the next step without further purification. An analytical sample was obtained by recrystallization in acetone; mp 79-80°C (lit. [11] 82-83°C).
3-Aminothiophenol (3)
In a 100-mL, 3-necked flask, fitted with a reflux condenser and a mechanical stirrer were placed 3-nitrophenyl disulfide (6.2 g, 20 mmol), iron powder (6.8 g, 122 mol) and 50% wt. aq EtOH (20 g). The mixture was stirred followed by the addition of concd HCl (0.4 mL) via a syringe. The resultant mixture was heated at reflux for 3 h, turned rusty and later, brown. It was then vacuum filtered while hot and the iron powder was returned to reaction flask and refluxed in 95% EtOH (20 mL) for 10 min and filtered hot. This step was repeated one more time. The combined filtrates was evaporated under reduced pressure to obtain the crude 3-aminothiophenol (3.7 g, 73%) as a liquid. The crude was used without further purification. It was identified by retention time comparison with a commercial sample from Aldrich.
3-[(3-Aminophenyl)disulfanyl]aniline (4) (Method A)
To a mixture of 3-nitrophenyl disulfide (25 g, 0.081 mol), iron powder (27.2 g, 0.488 mol) and 50% wt. aq EtOH (80 mL) in a 250-mL, round-bottomed flask was carefully added a mixture of concd HCl (17.6 g, 0.178 mol) and 50% wt. aq EtOH (34 mL) via an addition funnel under mechanical stirring and nitrogen in a period of 30 min. The reaction temperature was kept between 20-35°C with an ice-water bath until the completion of the reaction. The reaction mixture was then diluted with 95% EtOH (34 mL) and stirred for an additional 20 min. The dark brown mixture was then vacuum-filtered, rinsed with 95% EtOH (2x34 mL) and discarded. The filtrate was then poured into 6 N H2SO4 (34 mL) under stirring to afford a white slurry which was filtered under vacuum and washed with H2O (50 mL). The crude product was dried in a vacuum oven at 50°C with N2 purge to a constant weight of 29.8 g (106%).
The crude was used for next step without further purification.
3-[(3-Aminophenyl)disulfanyl]aniline (4) (Method B)
A mixture of 3-aminothiophenol (3) (4.8g, 38.3 mmol) and methyl sulfoxide (3.0 g, 38.3 mmol) was heated at 80-90°C in an oil bath for 4 h. GC analyses showed a clean conversion to 3-aminophenyl disulfide. The hot reaction mixture was then rapidly poured into a 35% H2SO4 soln (28 mL) in a beaker with stirring to obtain 3-aminophenyl disulfide sulfate as a white suspension which can be used for a diazonium reaction directly.
3-[(3-Hydroxyphenyl)disulfanyl]phenol (5)
3-Aminophenyl disulfide sulfate (42 g, 0.123 mol) was dissolved in concd H2SO4 (330 g) in a 600-mL beaker (beaker 1). Sodium nitrite powder (17.6 g, 0.255 mol) was then added to the beaker in several small portions so that the reaction temperature could be kept below 20°C with an ice-water bath. The viscous liquid was mechanically stirred for additional 10 min after completion of addition and was kept cold.
In a separate 1000-mL beaker (beaker 2) was placed crushed ice (300 g) and the beaker was immersed in an ice-water bath. To this beaker was then carefully poured the above diazonium soln in beaker 1 while the internal temperature was maintained below 15°C. The beaker 1 was washed with ice (2x15 g) and the resultant ice-water mixture was transferred into beaker 2. The beaker 2 was stirred for 10 additional min. Sulfamic acid (3.0 g) crystals were then added, the resultant mixture was stirred for additional 20 min and kept cold.
A 3000-mL, 3-necked, round-bottomed flask equipped with a mechanical stirrer, an addition funnel and a Claisen adapter through which were fitted a thermal couple and a condenser. In the flask were placed H2O (300 g), concd H2SO4 (300 g) and the resultant soln were heated to reflux using a heating mantle. The diazonium salt soln was then added to the refluxing sulfuric acid at such rate that the internal temperature could be kept between 120-124°C. Heating was removed shortly after the completion of addition and the flask was cooled to ca. 50°C with an ice-water bath followed by addition of toluene (600 mL). The mixture was heated at 50-70°C for 30 min under vigorous stirring and allowed to cool to r.t. The mixture was then transferred to a 2 L separatory funnel to separate the organic layer (the black tar was discarded). The aq layer was returned to the flask and extracted with toluene (2 × 600 mL). The combined toluene layer was dried over anhyd Na2SO4 and evaporated under reduced pressure to afford the crude 3-hydroxyphenyl disulfide (22.5 g, 75% yield) as a semi-solid. The crude was used without purification. The compound was identified by GCMS and HPLC by comparing with an authentic sample prepared by oxidation of 3-hydroxythiophenol with DMSO.
3-Hydroxythiophenol
The crude 3-hydroxyphenyl disulfide (66.3 g, 0.265 mol) was dissolved in anhyd EtOH (200 mL) in a 1 L, 3-necked, round-bottomed flask equipped with a mechanical stirrer and a thermal couple. To the flask was then added 10% wt. H2SO4 (300 g, 0.306 mol) to obtain a yellowish suspension. Zinc dust (17.3 g, 0.265 mol) was then added to the flask in several portions. The internal temperature was kept at 25-40°C for 1 h after the addition. The mixture was then heated at reflux to destroy any residual visible zinc particles. EtOH was stripped off at reduced pressure (230 mL liquid was collected). The residue was extracted with toluene (3x250 mL). The combined toluene layers were washed with brine (200 mL) and dried over anhyd Na2SO4. Removal of solvent on a rotary evaporator afforded crude 3-hydroxythiophenol as a liquid. It was purified by fractional vacuum distillation to yield pure 3-hydroxythiophenol (34.7 g, 54%) as a colorless liquid (92°C/0.075 mmHg). It was shown identical to that made via the Grignard route by 1H NMR, 13C NMR, IR and GCMS.