Abstract
A process for producing oxymorphone by converting oxycodone utilizing a suitable boron reagent in the presence of a weak Lewis base attenuating agent.
Process for Producing Oxymorphone
This invention relates to a process for producing oxymorphone.
Oxymorphone is a narcotic substance. It is widely used as an analgetic. The most frequently used method for producing oxymorphone is described by Seki, Takamine Kenkyisho Nempo, 12, 52 (1960): It involves reacting pyridine hydrochloride with oxycodone at high temperatures. It is disadvantageous on a commercial scale because the reaction is difficult to control, and high temperatures are needed. Further, this reaction produces only moderate yields along with the formation of substantial amounts of by-products.
It is an object of this invention to provide a process for producing oxymorphone in good yields with substantially no by-products.
It is a further object of the present invention to selectively demethylate the oxycodone methoxy group without affecting the other sites in the oxycodone molecule where ether linkages can be cleaved. In accordance with this invention, there is provided a process for producing oxymorphone by selectively removing the methyl group from the methoxy group of oxycodone comprising reacting oxycodone with a demethylating amount of a demethylating agent under demethylating conditions in the presence of an attenuating amount of an attenuating agent to attenuate the activity of the demethylating agent, whereby oxymorphone is produced in good yields with substantially no by-products.
Suitable demethylating agents are boron compounds capable of demethylating the methoxy group but incapable of forming numerous by-products. Such boron compounds include boron tribromide, boron trichloride or the reaction product of such halides with alcohols, e.g., those containing 1 to 10 carbon atoms, preferably lower alcohols such as those containing 1 to 6 carbon atoms, e.g., methanol, propanol, butanol, hexanol, etc.
Present in the reaction medium during the demethylating reaction is an attenuating agent-to attenuate the activity of the boron compound such that when utilized in the process of this invention,. good yields of oxymorphone are produced with substantially no by-products. The attenuating agent can be a weak Lewis base which does not chemically react with the demethylating agent. Attenuating agents include normally liquid aromatic solvents that do not chemically react with the boron compound, e.g., benzene, toluene, xylene, ethylbenzene, nitrobenzene, chlorobenzene, diphenyl ether and mixtures thereof. Chlorobenzene is the preferred attenuating agent. Attenuating amounts include from 25% to 900% by weight based on the weight of the boron compound.
It is preferred to utilize a demethylating composition which contains a boron compound in an amount sufficient to demethylate the methoxy group of oxycodone, e.g. from about 5 to 20% preferably about 10% based on the total weight of the demethylating composition and an attenuating amount of an attenuating agent, e.g., 80 to 95% preferably about 90% by weight, based on the total weight of the demethylating composition.
Oxymorphone is reacted with the aforesaid demethylating agent under demethylating conditions. This includes using a demethylating amount of the demethylating agent, e.g., in the case of boron trihalide, from about 2 to 8 moles, advantageously 2.5 to 3.5 moles, preferably 2.5 to 7 moles, of the boron compound per mole of oxycodone. No significant advantage is achieved by using more than 8 moles, although this is possible. Utilizing less than about 2 moles may result in an incomplete reaction. Other demethylating conditions include suitable reaction times, e.g., 8 to 24 hours and reaction temperatures, e.g., from about 0 to 40°C. As mentioned, it is preferred to react oxycodone with the aforesaid demethylating composition. Normally, the demethylating composition is sufficiently fluid that no further solvent is necessary to carry out the reaction. However, it may be advantageous to add a solvent, e.g., an inert solvent that will not react with the boron compound, e.g., chlorobenzene. Such a solvent is preferably the same as but can be different from the attenuating agent employed. Alternatively, the demethylating agent can be added to the reaction medium separately, provided the attenuating agent is present in a sufficient amount to attenuate the activity of the demethylating agent. For example, oxycodone can be mixed with the attenuating agent to which is added the demethylating agent.
After oxycodone demethylation has occurred to the extent desired, the demethylation reaction is quenched by adding to the reaction medium a quenching amount of water. Advantageously, water is added in an amount equal to or greater than the volume of the anhydrous reaction medium.
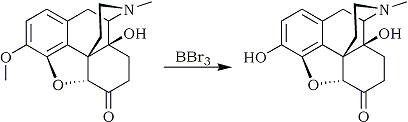
To maximize yield of oxymorphone, the quenched reaction mixture is advantageously hydrolyzed for a period of time and under hydrolysis conditions sufficient to increase the amount of recoverable oxymorphone present in the reaction medium. Hydrolysis serves to hydrolyze both excess reactants and reaction products present in the reaction medium after demethylation. Suitable hydrolysis step reaction times include hydrolysis for from about 1/2 to 10 hours, preferably from 2 to 4 hours. Suitable temperatures for the hydrolysis step range from about 60°C to 120°C, preferably from about 80°C to 100°C. It has been found that hydrolyzing the reaction mixture at higher temperatures, e.g. at the reflux temperature of the reaction mixture, is especially advantageous, particularly when the preferred chlorobenzene attenuating agent/solvent is employed. Without being bound by any particular theory, it is believed that hydrolysis at higher temperatures promotes hydrolysis of those reaction products in the form of boron complexes such as complexes containing a boron- nitrogen bond, thereby converting more of such complexes into recoverable oxymorphone. Higher hydrolysis temperatures may also serve to convert other reaction by-products present into recoverable product. After hydrolysis, the pH of the reaction mixture is adjusted to about 4.5 to about 6 with acid, e.g., hydrochloric or sulfuric, filtered and then adjusted with a suitable base, e.g., sodium hydroxide, to a pH of about 10 to 12 and extracted with one of the conventional inert organic extraction solvents, e.g. toluene. The aqueous layer is then adjusted to a pH of about 2 with acid and then to about 8.5 with base and extracted with an inert organic extraction solvent, e.g., methylene chloride, which is evaporated to give oxymorphone substantially free of impurities. The first organic extract is evaporated to give oxycodone suitable for recycle.
The following examples illustrate the invention. All parts are by weight unless otherwise stated.
Example 1
Table I |
|||
| Run # | Solvent | Moles BBr3 | Yield |
| 1 | Chlorobenzene | 3 | 76% |
| 2 | Toluene | 3 | 82%a |
| 3 | CHCl3 | 4 | 66% |
| 4 | CH2Cl2 | 4 | 51% |
| 5 | CHCl3 | 6 | 51% |
| 6 | Dichloroethane | 4 | 29% |
| 7 | sym-Tetrachloroethane | 4 | 65% |
a. 9% Oxycodone recovered
A slurry of 25g of oxycodone base in 200ml of chlorobenzene is placed in a vessel equipped for efficient stirring and the contents are cooled to less than 10°C. A solution of 60g of boron tribromide is prepared in 200ml of chlorobenzene and this solution is added over a span of 5 minutes to the oxycodone slurry. The temperature rises to about 35°C. The cooling is removed and the mixture is stirred for 18 hours. At this point the mixture is poured into 250ml of water and the mixture is refluxed for 2 hours. The aqueous and organic layers are separated and the aqueous layer is assayed for oxycodone and oxymorphone. The aqueous layer is adjusted to a pH of 5.5 with sodium hydroxide or ammonia and filtered. The filtrate is adjusted to a pH of 12 with sodium hydroxide and exhaustively extracted with methylene chloride. The methylene chloride layer is separated and evaporated to give oxycodone which can be recycled. The aqueous layer is acidified with hydrochloric acid to a pH of 2.0 and then adjusted to a pH of 8.5 with ammonia and exhaustively extracted with methylene chloride. The organic layer is evaporated to give substantially pure oxymorphone.
Data for this experiment are shown in the following Table I as well as that obtained for other solvents following substantially the same procedure.
Example 2
Table II |
||||
| Run # | Solvent | Moles BBr3 | Yield | Oxycodone recovered |
| 1 | CHCl3/Toluene 1:1 | 4 |
70% | |
| 2 | Toluene | 4 |
76% | 15% |
| 3 | Toluene | 6 |
50% | |
| 4 | Toluene | 3 |
85% | 5% |
| 5 | Toluene | 2 |
62% | 10% |
| 6 | Toluene | 1 |
23% | 68% |
| 7 | Xylene | 3 |
81% | 6% |
| 8 | Chlorobenzene | 3 (BCl3) |
70% | |
A slurry of 1.5 g of oxycodone in benzene is treated all at once with 2.3g of boron tribromide in benzene and the mixture stirred for 2 hours, the mixture was hydrolyzed with an equal volume of water at reflux for 2 hours. The aqueous layer was assayed to indicate an 85% yield of oxymorphone and a 15% yield of oxycodone. By essentially following the procedure of Example 2 the data in Table II was obtained.
Example 3
Using the following general procedures, oxycodone is demethylated to form oxymorphone using a boron tribromide demethylating agent. Variations in boron tribromide/oxycodone ratio, type of reaction medium organic solvent employed and hydrolysis conditions are set forth in Table III:
| Table III Oxycodone to Oxymorphone, Conversion |
||||||
| Run No. | BBr3 | Oxycodone | BBr3:Oxycodone (Moles) |
Solvent | Hydrolysis Temp. | Yield of Oxymorphone |
| 1 | -75g |
-15g |
6 |
CHCl3 |
40°C |
66% |
| 2 | 71g |
15g |
6 |
CHCl3 |
reflux |
71% |
| 3 | 35g |
15g |
3 |
CHCl3 |
40°C |
51% |
| 4 | 35g |
15g |
3 |
CHCl3 |
reflux |
67% |
| 5 | 75g |
15g |
6 |
PhCl |
reflux |
88% |
| 6 | 60g |
25g |
3 |
PhCl |
reflux |
85% |
| 7 | 75g |
15g |
6 |
PhCl |
40°C |
70% |
| 8 | 78g |
15g |
3 |
PhCl |
40°C |
72% |
Boron tribromide in solvent is added to a slurry of oxycodone in solvent for Runs 5-8 with the reaction medium temperature being maintained below about 10°C. The order of addition is reversed for Runs 1-4. After reaction times varying from 1 to 20 hours, the reaction mixture is quenched by addition of water. Hydrolysis of the reaction mixture then takes place either at 40°C or at reflux temperature of the reaction medium. The pH of the reaction mixture is then adjusted to about 5.5, and the reaction mixture is analyzed for oxymorphone conversion using liquid chromatography techniques.
The above Table III data illustrate that oxymorphone yield is improved by utilizing the chlorobenzene solvent/attenuating agent and by utilizing hot hydrolysis conditions.
Example 4
Approximately 2.68kg of oxycodone are added to a 189 liter reaction vessel furnished with a heating/cooling jacket, said vessel containing 48kg of chlorobenzene. The contents of the reaction vessel are rapidly stirred and the system is purged with nitrogen. About 7.8kg of boron tribromide are added to the mixture over a period of 20-30 minutes during which time the temperature of the reaction mixture is kept below 25°C.
Upon completion of the boron tribromide addition, the contents of the reaction vessel are stirred for 6 hours at room temperature (25-28°C). At this point the reaction mixture is pumped with stirring to a 246 liter vessel containing 32.66kg of water which has been cooled to less than 10°C. The addition process is such that the vessel temperature remains below 30°C.
The resulting slurried mixture is pumped back into the 189 liter reaction vessel and is heated to reflux (96°C) with slow stirring so as to prevent emulsion formation. After two hours of refluxing, the reaction vessel contents are cooled to 60-80°C and the layers are allowed to separate. Upon separation the bottom aqueous layer is removed and the organic layer is rinsed with 5.67kg of de-ionized water and slowly stirred. As the mixture settles, the layers switch so that the aqueous layer becomes the top layer., The aqueous layer is removed and combined with the previous aqueous extraction. The pH of the organic layer is adjusted to 5.5- 6.0 with ammonium hydroxide. About 0.45-0.9kg of Darco (activated carbon; Darco G-60) are added and the resulting mixture is filtered and then washed with 3.8-7.6 liters of de-ionized water. The pH is readjusted to 8.8-8.9 with ammonium hydroxide. The resulting aqueous slurry is extracted with dichloromethane in a continuous Karr column extractor until the aqueous portion contains less than 1.5 mg of oxymorphone per ml. The dichloromethane portion is back-washed with 2 3.8 liter portions of de-ionized water and' is returned to a 189 liter reaction vessel having a heating/cooling jacket which is maintained at 70-80°C.
The dichloromethane solution is stripped to dryness and the last of the dichloromethane and residual water is removed under vacuum. About 30 liters of anhydrous ethanol are added to the reaction vessel whereupon the resulting mixture is warmed to 65-70°C. The mixture is filtered if necessary and the ethanol mixture is then cooled with stirring to less than 10°C. The resulting crystallization product is filtered and dried at 65-75°C for 2-4 hours to give 1.36-1.59kg of oxymorphone.
The filtrate is stripped to near dryness and is replaced with 7.57 liters of de-ionized water. The pH is adjusted to less than 5.0 and is subsequently readjusted to 8.5-8.8 with ammonium hydroxide while maintaining the temperature of the mixture at less than 30°C. The mixture is cooled to 10-15°C and the resulting precipitated solid is filtered and washed with two 0.95 liter portions of water at 10-15°C. The solid is dried at 70-80°C for a minimum of 6 hours to give 0.36 to 0.5kg of residue as additional oxymorphone-containing product.
Example 5
Preparation with Oxymorphone with Recovery of Unreacted Oxycodone
Oxycodone (50g) in 400ml of chlorobenzene is cooled to 8°C and treated with 120g of BBr3 in 400ml of chlorobenzene, over 10 minutes. The reaction medium is stirred for one hour at room temperature and is then added to 500 ml of ice water. The mixture is heated to reflux. The aqueous layer is then cooled and separated, and adjusted to a pH of 5.5 with ammonia. Activated charcoal is added, and the liquid is filtered with Celite. The pH is then adjusted to 8.5 with ammonia. The aqueous layer is then exhaustively extracted with methylene chloride. The methylene chloride layer is extracted with diluted sodium hydroxide solution. The aqueous layer is then adjusted to a pH of 4 and then 8.5, and the precipitated oxymorphone is collected by filtration. The methylene chloride layer is extracted with 1N HCl. The resulting aqueous layer is treated with ammonia to give a precipitate of oxycodone.
Altogether, 27.2g of oxymorphone and 7.1g of oxycodone are recovered.