Iodination and Hydroxylation of Aromatic CompoundsUnited States Patent 4,465,864[ Back to the Chemistry Archive ] 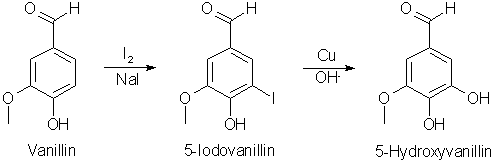
Lignin derived aromatic compounds are frequently inexpensive precursors of a number of valuable organic substances. For example, vanillin is an important precursor chemical of 3,4,5-trimethoxybenzaldehyde, a chemical of well known importance to the pharmaceutical industry. However, known processes for the conversion of vanillin to trimethoxybenzaldehyde have certain drawbacks. U.S. Pat. No. 3,855,306 describes one such process in which vanillin is brominated to produce 5-bromovanillin using bromine and a concentrated hydrobromic acid solvent. The resulting 5-bromovanillin is then isolated from the reaction mixture and hydrolyzed to the corresponding hydroxyvanillin with sodium hydroxide and a copper catalyst. The resulting reaction mixture includes sodium bromide which is normally not recycled but rather is discarded or used elsewhere. Moreover, bromine and hydrobromic acid are both highly corrosive and hazardous to handle. Other halogenated vanillin derivatives include 5-chlorovanillin and 5-iodovanillin. It is known however that chlorovanillin is unreactive toward hydroxide ion/copper and yields no hydroxyvanillin using conditions even more drastic than those effective for converting 5-bromovanillin to 5-hydroxyvanillin. Aryl iodides, on the other hand, are known to be more reactive than the corresponding bromocompounds in this kind of reaction. Iodine, however, would be prohibitively expensive if discarded as the bromine is in the above described bromination reaction. Reference to the iodination of vanillin to 5-iodovanillin may be found in H. Erdman, Svensk. Kem. Tids., 47, 223 (1935) Chem. Abstr. 30:449. Conversion of 5-iodovanillin to 5-hydroxyvanillin is disclosed in S. Banerjee, M. Manolopoulo and J. M. Pepper, Canadian Journal of Chemistry, 40, 2175 (1962). However, the processes there disclosed isolate the intermediate iodovanillin before hydrolysis and do not recover the by-product iodide ion from the various reactions. A process has now been discovered from the conversion of armoatic compounds to hydroxy aromatic compounds which does not isolate an intermediate haloaromatic compound and which permits economical recovery and reuse of halide by-products produced during the reaction. The process involves the hydroxylation of an aromatic compound by reacting the aromatic compound in the presence of an aqueous solvent with a triiodide salt to form a reaction mixture containing the corresponding iodoaromatic compound, reacting the mixture, without separation of the iodoaromatic compound with a hydroxylating agent to form the corresponding hydroxy aromatic compound and additional iodide salt, separating the hydroxy aromatic compound from the iodide salt and recapturing the iodide salt. The corresponding alkoxy aromatic compound may be produced by alkylation of the hydroxy aromatic compound by known alkylation procedures. The process of the invention renders cost-effective an otherwise cost-ineffective process using the iodination route. In the case of such processes as the conversion of vanillin to trimethoxybenzaldehyde, the process becomes basically a one-step or "one-pot" process by eliminating the purification of the intermediate 5-iodovanillin. Iodination of vanillin and conversion of the resulting iodovanillin to hydroxyvanillin may be carried out in the same reaction vessel. Moreover, efficient recycle of the by-product iodide salt to the triiodide reagent used in the reaction obviates the need to dispose of the valuable iodine/iodide material. Prior art processes necessarily separated and recovered the bromide or iodide by-product from the halogenation reaction and again from the hydroxylation reaction. In the present process, the iodide salt is recovered only after hydroxylation, at which time it may be oxidized to iodine and the iodine partially reduced to form the starting triiodide reactant. (The triiodide reagent is a solution of iodine in an excess of the iodide salt, e.g., NaI+I2 or NaI3). If chlorine is used as the oxidizing agent, the only net by-product of the reaction is sodium chloride, an obviously inexpensive waste by-product. The starting materials useful in the practice of the invention are aromatic compounds subject to electrophilic substitution reactions. Such compounds may include benzene but the process is particularly suitable for aromatic compounds containing electron donating nuclear substituents, i.e. mono- or polycyclic aromatic compounds containing one or more hydrocarbon substituents such as an alkyl, cycloalkyl, aryl or aralkyl group and/or one or more hydroxy groups or aldehyde, acid, ester or ether radicals, i.e., alkoxy, carboxy, carboxyl or aldehyde carbonyl groups. The process is not useful with substituents such as poly-nitro, or ketone groups with an alpha hydrogen, which either react with the reagent or strongly de-activate the ring. Ketone groups are deactivating, as are aldehyde groups, but ketones containing an alpha hydrogen would react with the iodinating agent whereas the aldehydes would not. For example, diaryl ketones would not interfere. Only in the case of severely de-activating groups, such as poly-nitro groups, is the de-activation a problem in carrying out the iodination. Weakly de-activating groups such as aldehyde groups do not interfere with iodination. Useful aromatic compounds are simple monohydric phenols such as phenol, o-, m- and p-cresol and guaiacol; polyhydric phenols such as catechol and resorcinol; phenolic aldehydes such as protocatechualdehyde, vanillin, syringaldehyde, p-hydroxybenzaldehyde and 5-formylvanillin; phenolic acids such as vanillic acid, syringic acid, protocatechuic acid and p-hydroxybenzoic acid. The preferred aromatic reactants are those having at least one phenolic hydroxyl functionality. The first step of the reaction involves iodination of the aromatic compound with the triiodide salt in the presence of water as a solvent. The water should contain from 0.7 to 1.25 molar equivalents of a hydroxide, preferably an alkali metal hydroxide, and from 1-2 molar equivalents of an alkali metal triiodide (e.g. iodine plus sodium iodide). The aqueous solvent should also contain from 0.1 to 20 mole % of an acid catalyst, which may be a mineral acid such as sulfuric, hydrochloric or phosphoric acid. Reaction is carried out at temperatures ranging from 20-120°C. If the starting compound contains a nuclear substituent, iodination will occur in the ortho or para position on the nuclear ring. The subsequent step of the reaction, hydroxylation, is carried out directly with the reaction mixture from iodination without any intermediate isolation or other processing of the reactants or by-products. A base, such as an alkali metal hydroxide or a quaternary amine such as tetraalkylammonium hydroxide, is added directly to the reaction mixture to make a final concentration of 0.5 to 6 molar, with 0.1 to 20 mole % copper metal, or cuprous salts such as oxide, chloride or iodide, at temperatures of from 50-120°C. The preferred conditions are addition of sodium hydroxide to the iodination reaction mixture to give a concentration of 2-5 molar, then addition of 1-5 mole % copper dust, cuprous oxide or cuprous chloride, then allowing reaction at reflux (100-120°C.) for about 18 hours. The sodium or other iodide ion by-product may be recovered by neutralizing the caustic in the reaction mixture with an acid such as sulfuric or hydrochloric, extracting the organic product from the water solvent and then treating the water solution with an oxidizing agent. The oxidizing agent may be chlorine, sodium hypochlorite, hydrogen peroxide, persulfate, perborate or electrochemical oxidation may be used. The iodine which precipitates is then recovered from the water solvent by filtration, solvent extraction or distillation/sublimation. The temperature of the water phase may be from 0-100°C. The preferred method for iodine recovery is treatment of the water solution with sulfuric acid to neutralize the base, extraction of the hydroxy aromatic compound with organic solvents such as methylene chloride or toluene, oxidation with chlorine or electrochemically and filtration or solvent extraction to recover the iodine. The crude hydroxy aromatic compound may then be used directly in any subsequent alkylation procedure. A specific description of a preferred practice of the invention with vanillin as the aromatic compound is as follows. Vanillin is dissolved in water with one molar equivalent of sodium hydroxide while the solution is warmed to 50-100°C. One molar equivalent of iodine and two molar equivalents of sodium iodide are added to water to prepare one molar equivalent of NaI3.NaI. This sodium triiodide solution is added to the sodium vanillate solution along with a catalytic amount of sulfuric acid--preferably from 5-10 mole %. The mixture is stirred about one hour at a temperature of 50-100°C, then sodium hydroxide is added to make the solution alkaline (from 1 to 5N). The copper catalyst is then added and the mixture heated at reflux until the iodovanillin is consumed, about 12 hours. The excess hydroxide is then neutralized and the 5-hydroxyvanillin extracted with a water-immiscible organic solvent. The aqueous phase bearing the sodium iodide is then subjected to oxidizing conditions and the resultant iodine precipitates from solution. The solid element is filtered out, and a sodium triiodide solution prepared by reducing a portion of the iodine to sodium iodide and dissolving the iodine in the iodide to make the sodium triiodide solution. Alkylation of the hydroxy aromatic compound to the corresponding alkoxy aromatic compound may be performed in accordance with known alkylation procedures in which the hydroxy aromatic compound is reacted with an alkyl sulfate, alkyl halide or alkyl sulfonate in a suitable solvent, usually water, containing a base such as sodium hydroxide. Such reactions are shown at various places in the literature, as for ex. in Organic Synthesis, Col. Vol. II, page 619, 1943, in which veratraldehyde is prepared from vanillin. The iodide salt may, if desired, be recaptured subsequent to the alkylation reaction. The following examples illustrate the practice of the invention. Unless otherwise indicated, all parts and percentages are by weight.
The process thus provides an essentially one-step process for the nearly quantitative conversion of aromatic compounds to hydroxy aromatic compounds and for the recovery and recycle of the reagent used for conversion. 5-Iodovanillin (JCS 3740-3741 (1958)) 12.6g Iodine was added in 4 portions during 30 min to a rapidly stirred suspension of 7.5g vanillin in 200mL H2O containing 5g NaHCO3 and 10g KI. Stirring was continued for 3h and the mixture left overnight. The filtered product was washed with dilute Na2S2O3 and H2O and dried at 45°C (11.8g, mp 175°C). Crystallised from EtOH(aq), it had mp 180°C. |